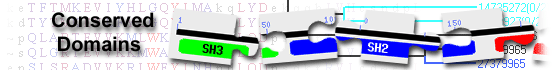|
Name |
Accession |
Description |
Interval |
E-value |
| Filament |
pfam00038 |
Intermediate filament protein; |
145-458 |
7.81e-151 |
|
Intermediate filament protein;
Pssm-ID: 459643 [Multi-domain] Cd Length: 313 Bit Score: 434.73 E-value: 7.81e-151
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 145 EEREQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQGTKTVRqNLEPLFEQYINNLRRQLDSIVGERGRLDSELRN 224
Cdd:pfam00038 1 NEKEQLQELNDRLASYIDKVRFLEQQNKLLETKISELRQKKGAEPS-RLYSLYEKEIEDLRRQLDTLTVERARLQLELDN 79
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 225 MQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLRALYDAELSQMQTHISDTSVVLS 304
Cdd:pfam00038 80 LRLAAEDFRQKYEDELNLRTSAENDLVGLRKDLDEATLARVDLEAKIESLKEELAFLKKNHEEEVRELQAQVSDTQVNVE 159
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 305 MDNNRNLDLDSIIAEVKAQYEEIAQRSRAEAESWYQTKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQ 384
Cdd:pfam00038 160 MDAARKLDLTSALAEIRAQYEEIAAKNREEAEEWYQSKLEELQQAAARNGDALRSAKEEITELRRTIQSLEIELQSLKKQ 239
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 194388850 385 CASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQELMNVKLALDVEIATYRKLLEGEECR 458
Cdd:pfam00038 240 KASLERQLAETEERYELQLADYQELISELEAELQETRQEMARQLREYQELLNVKLALDIEIATYRKLLEGEECR 313
|
|
| Keratin_2_head |
pfam16208 |
Keratin type II head; |
17-142 |
1.52e-26 |
|
Keratin type II head;
Pssm-ID: 465068 [Multi-domain] Cd Length: 156 Bit Score: 105.51 E-value: 1.52e-26
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 17 GFSANSARLPGVSRSGFSSISVSRSRGSGGLGGACGGAGFGSRSLYGLGGSKRISI----------------GGGSCAIS 80
Cdd:pfam16208 1 GFSSCSAVVPSRSRRSYSSVSSSRRGGGGGGGGGGGGGGFGSRSLYNLGGSKSISIsvagggsrpgsgfgfgGGGGGGFG 80
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 194388850 81 GGYGSRAGGSYGFGGAGSGFGLGGFGGPG--------------FPVCPPGGIQEVTVNQSLLTPLNLQIDPAIQRV 142
Cdd:pfam16208 81 GGFGGGGGGGFGGGGGFGGGFGGGGYGGGgfggggfggrggfgGPPCPPGGIQEVTVNQSLLQPLNLEIDPEIQRV 156
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
145-440 |
2.84e-11 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 66.62 E-value: 2.84e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 145 EEREQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQGTKTVRQNLEplFEQYINNLRRQLDSIVGERGRLDSELRN 224
Cdd:TIGR02168 674 ERRREIEELEEKIEELEEKIAELEKALAELRKELEELEEELEQLRKELEE--LSRQISALRKDLARLEAEVEQLEERIAQ 751
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 225 MQDLVEDLKNKYEDEVNKRTAAENEFVTLKKdvdaaymNKVELQAKADTLTDEINFLRALYD---AELSQMQTHISDTSV 301
Cdd:TIGR02168 752 LSKELTELEAEIEELEERLEEAEEELAEAEA-------EIEELEAQIEQLKEELKALREALDelrAELTLLNEEAANLRE 824
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 302 VLSMDNNR-------NLDLDSIIAEVKAQYEEIAQ----------RSRAEAESWYQtKYEELQVTAGRHGDDLRNTKQEI 364
Cdd:TIGR02168 825 RLESLERRiaaterrLEDLEEQIEELSEDIESLAAeieeleelieELESELEALLN-ERASLEEALALLRSELEELSEEL 903
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 365 AEINRMIQRLRSEIDHVKKQCASLQAAIADAEQR-----------GEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQE 433
Cdd:TIGR02168 904 RELESKRSELRRELEELREKLAQLELRLEGLEVRidnlqerlseeYSLTLEEAEALENKIEDDEEEARRRLKRLENKIKE 983
|
....*..
gi 194388850 434 LMNVKLA 440
Cdd:TIGR02168 984 LGPVNLA 990
|
|
| PRK09039 |
PRK09039 |
peptidoglycan -binding protein; |
286-426 |
9.92e-08 |
|
peptidoglycan -binding protein;
Pssm-ID: 181619 [Multi-domain] Cd Length: 343 Bit Score: 54.20 E-value: 9.92e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 286 DAELSQMQTHISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEiAQRSRAEAESWYQTKYEELQVTAGRHGD---DLRNTKQ 362
Cdd:PRK09039 52 DSALDRLNSQIAELADLLSLERQGNQDLQDSVANLRASLSA-AEAERSRLQALLAELAGAGAAAEGRAGElaqELDSEKQ 130
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....*...
gi 194388850 363 EIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGemalKDAKNKLEG----LEDALQKAKQDLAR 426
Cdd:PRK09039 131 VSARALAQVELLNQQIAALRRQLAALEAALDASEKRD----RESQAKIADlgrrLNVALAQRVQELNR 194
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
128-417 |
1.91e-07 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 54.29 E-value: 1.91e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 128 LTPLNLQIDPAIQRVRAEEREqIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQgTKTVRQNLEpLFEQYINNLRRQ 207
Cdd:TIGR02168 241 LEELQEELKEAEEELEELTAE-LQELEEKLEELRLEVSELEEEIEELQKELYALANE-ISRLEQQKQ-ILRERLANLERQ 317
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 208 LDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLR---AL 284
Cdd:TIGR02168 318 LEELEAQLEELESKLDELAEELAELEEKLEELKEELESLEAELEELEAELEELESRLEELEEQLETLRSKVAQLElqiAS 397
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 285 YDAELSQMQTHISDTSVVLSMDNNRNLDLDSII--AEVKAQYEEIAQRSRAEaeswyqtkyEELQVTAGRHGDDLRNTKQ 362
Cdd:TIGR02168 398 LNNEIERLEARLERLEDRRERLQQEIEELLKKLeeAELKELQAELEELEEEL---------EELQEELERLEEALEELRE 468
|
250 260 270 280 290
....*....|....*....|....*....|....*....|....*....|....*..
gi 194388850 363 EIAEINRMIQRLRSEIDHVKKQCASLQAAIADAE--QRGEMALKDAKNKLEGLEDAL 417
Cdd:TIGR02168 469 ELEEAEQALDAAERELAQLQARLDSLERLQENLEgfSEGVKALLKNQSGLSGILGVL 525
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
204-453 |
2.76e-07 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 53.53 E-value: 2.76e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 204 LRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLRA 283
Cdd:TIGR02169 679 LRERLEGLKRELSSLQSELRRIENRLDELSQELSDASRKIGEIEKEIEQLEQEEEKLKERLEELEEDLSSLEQEIENVKS 758
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 284 L---YDAELSQMQTHISdtSVVLSMDNNRNLDLDSIIAEVKAQYEEI-AQRSRAEAeswyqtKYEELQVTAGRHGDDLRN 359
Cdd:TIGR02169 759 ElkeLEARIEELEEDLH--KLEEALNDLEARLSHSRIPEIQAELSKLeEEVSRIEA------RLREIEQKLNRLTLEKEY 830
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 360 TKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRgemaLKDAKNKLEGLEDALQKAKQDLARLLKEYQELMNVKL 439
Cdd:TIGR02169 831 LEKEIQELQEQRIDLKEQIKSIEKEIENLNGKKEELEEE----LEELEAALRDLESRLGDLKKERDELEAQLRELERKIE 906
|
250
....*....|....
gi 194388850 440 ALDVEIATYRKLLE 453
Cdd:TIGR02169 907 ELEAQIEKKRKRLS 920
|
|
| PRK01156 |
PRK01156 |
chromosome segregation protein; Provisional |
135-456 |
4.94e-07 |
|
chromosome segregation protein; Provisional
Pssm-ID: 100796 [Multi-domain] Cd Length: 895 Bit Score: 52.60 E-value: 4.94e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 135 IDP-AIQRVRAEEREQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQ------GTKTVRQNLEPLFEQYINNLRR- 206
Cdd:PRK01156 402 IDPdAIKKELNEINVKLQDISSKVSSLNQRIRALRENLDELSRNMEMLNGQsvcpvcGTTLGEEKSNHIINHYNEKKSRl 481
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 207 --QLDSIVGERGRLDSELRNMQDLVEDLKNKyedEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLRAL 284
Cdd:PRK01156 482 eeKIREIEIEVKDIDEKIVDLKKRKEYLESE---EINKSINEYNKIESARADLEDIKIKINELKDKHDKYEEIKNRYKSL 558
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 285 YDAELSQMQTHISDTSVVLSmdnnrNLDLDSIIA---EVKAQYEEIAQRSRaEAESWYQTKYEELQVTAGRHGDD---LR 358
Cdd:PRK01156 559 KLEDLDSKRTSWLNALAVIS-----LIDIETNRSrsnEIKKQLNDLESRLQ-EIEIGFPDDKSYIDKSIREIENEannLN 632
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 359 NTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMALkDAKNKLEGLEDALQKAKQDLARLLKEYQELMNVK 438
Cdd:PRK01156 633 NKYNEIQENKILIEKLRGKIDNYKKQIAEIDSIIPDLKEITSRIN-DIEDNLKKSRKALDDAKANRARLESTIEILRTRI 711
|
330
....*....|....*...
gi 194388850 439 LALDVEIATYRKLLEGEE 456
Cdd:PRK01156 712 NELSDRINDINETLESMK 729
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
174-434 |
7.41e-07 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 52.37 E-value: 7.41e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 174 LDTKWTLLQEQGTKTVRqnleplFEQYINNLRR-QLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVT 252
Cdd:TIGR02168 198 LERQLKSLERQAEKAER------YKELKAELRElELALLVLRLEELREELEELQEELKEAEEELEELTAELQELEEKLEE 271
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 253 LKKDVDAAYMNKVELQAKADTLTDEINFL---RALYDAELSQMQTHISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEIAQ 329
Cdd:TIGR02168 272 LRLEVSELEEEIEELQKELYALANEISRLeqqKQILRERLANLERQLEELEAQLEELESKLDELAEELAELEEKLEELKE 351
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 330 RSRA------EAESWYQ---TKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQR-G 399
Cdd:TIGR02168 352 ELESleaeleELEAELEeleSRLEELEEQLETLRSKVAQLELQIASLNNEIERLEARLERLEDRRERLQQEIEELLKKlE 431
|
250 260 270
....*....|....*....|....*....|....*
gi 194388850 400 EMALKDAKNKLEGLEDALQKAKQDLARLLKEYQEL 434
Cdd:TIGR02168 432 EAELKELQAELEELEEELEELQEELERLEEALEEL 466
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
187-434 |
7.46e-07 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 52.38 E-value: 7.46e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 187 KTVRQNLEpLFEQYINNLRRQLDSIVGERgrldSELRNMQDLVEDL-KNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKV 265
Cdd:TIGR02169 180 EEVEENIE-RLDLIIDEKRQQLERLRRER----EKAERYQALLKEKrEYEGYELLKEKEALERQKEAIERQLASLEEELE 254
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 266 ELQAKADTLTDEINFLRALydaeLSQMQTHISDtsvvlsMDNNRNLDLDSIIAEVKAqyeEIAQRSRAEAEswYQTKYEE 345
Cdd:TIGR02169 255 KLTEEISELEKRLEEIEQL----LEELNKKIKD------LGEEEQLRVKEKIGELEA---EIASLERSIAE--KERELED 319
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 346 LQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGE----------MALKDAKNKLEGLED 415
Cdd:TIGR02169 320 AEERLAKLEAEIDKLLAEIEELEREIEEERKRRDKLTEEYAELKEELEDLRAELEevdkefaetrDELKDYREKLEKLKR 399
|
250
....*....|....*....
gi 194388850 416 ALQKAKQDLARLLKEYQEL 434
Cdd:TIGR02169 400 EINELKRELDRLQEELQRL 418
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
201-434 |
1.09e-06 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 51.61 E-value: 1.09e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 201 INNLRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYE------DEVNKRTAA--ENEFVTLKKDVDAAYMNKVELQAKAD 272
Cdd:TIGR02169 232 KEALERQKEAIERQLASLEEELEKLTEEISELEKRLEeieqllEELNKKIKDlgEEEQLRVKEKIGELEAEIASLERSIA 311
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 273 TLTDEINFL---RALYDAELSQMQTHISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEIAQR----------------SRA 333
Cdd:TIGR02169 312 EKERELEDAeerLAKLEAEIDKLLAEIEELEREIEEERKRRDKLTEEYAELKEELEDLRAEleevdkefaetrdelkDYR 391
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 334 EAESWYQTKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQrgemalkdaknKLEGL 413
Cdd:TIGR02169 392 EKLEKLKREINELKRELDRLQEELQRLSEELADLNAAIAGIEAKINELEEEKEDKALEIKKQEW-----------KLEQL 460
|
250 260
....*....|....*....|.
gi 194388850 414 EDALQKAKQDLARLLKEYQEL 434
Cdd:TIGR02169 461 AADLSKYEQELYDLKEEYDRV 481
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
262-462 |
1.43e-06 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 51.21 E-value: 1.43e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 262 MNKVELQAKADT----LTDEINFL-RALYDAELSQMQTHISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEIaQRSRAEAE 336
Cdd:TIGR02168 202 LKSLERQAEKAErykeLKAELRELeLALLVLRLEELREELEELQEELKEAEEELEELTAELQELEEKLEEL-RLEVSELE 280
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 337 SwyqtKYEELQvtagrhgDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMA---LKDAKNKLEGL 413
Cdd:TIGR02168 281 E----EIEELQ-------KELYALANEISRLEQQKQILRERLANLERQLEELEAQLEELESKLDELaeeLAELEEKLEEL 349
|
170 180 190 200 210
....*....|....*....|....*....|....*....|....*....|..
gi 194388850 414 EDALQKAKQDLARLLKEYQELMNVKLALDVEIATYRK---LLEGEECRLNGE 462
Cdd:TIGR02168 350 KEELESLEAELEELEAELEELESRLEELEEQLETLRSkvaQLELQIASLNNE 401
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
214-460 |
2.04e-06 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 50.83 E-value: 2.04e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 214 ERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLRALYDAELSQMQ 293
Cdd:TIGR02168 678 EIEELEEKIEELEEKIAELEKALAELRKELEELEEELEQLRKELEELSRQISALRKDLARLEAEVEQLEERIAQLSKELT 757
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 294 THISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEIAQRSRA-----EAESWYQTKYEELQVTAGRHGDDLRNTKQEIAEIN 368
Cdd:TIGR02168 758 ELEAEIEELEERLEEAEEELAEAEAEIEELEAQIEQLKEElkalrEALDELRAELTLLNEEAANLRERLESLERRIAATE 837
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 369 RMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLE----------GLEDALQKAKQDLARLLKEYQELMNVK 438
Cdd:TIGR02168 838 RRLEDLEEQIEELSEDIESLAAEIEELEELIEELESELEALLNerasleealaLLRSELEELSEELRELESKRSELRREL 917
|
250 260
....*....|....*....|..
gi 194388850 439 LALDVEIATYRKLLEGEECRLN 460
Cdd:TIGR02168 918 EELREKLAQLELRLEGLEVRID 939
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
198-473 |
3.66e-06 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 49.63 E-value: 3.66e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 198 EQYIN-NLRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDevnkrTAAENEFVTLKKDVDAAYMNKVELQAKADTLTD 276
Cdd:COG3206 159 EAYLEqNLELRREEARKALEFLEEQLPELRKELEEAEAALEE-----FRQKNGLVDLSEEAKLLLQQLSELESQLAEARA 233
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 277 EINFLRALYDAELSQMQTHISDTSVVLSmdnnrnldlDSIIAEVKAQYEEIaqrsraeaeswyQTKYEELQVTAGRHGDD 356
Cdd:COG3206 234 ELAEAEARLAALRAQLGSGPDALPELLQ---------SPVIQQLRAQLAEL------------EAELAELSARYTPNHPD 292
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 357 LRNTKQEIAEINRMIQRLRSEIDhvkkqcASLQAAIADAEQRgemalkdaKNKLEGLEDALQKAKQDLARLLKEYQELMN 436
Cdd:COG3206 293 VIALRAQIAALRAQLQQEAQRIL------ASLEAELEALQAR--------EASLQAQLAQLEARLAELPELEAELRRLER 358
|
250 260 270 280
....*....|....*....|....*....|....*....|
gi 194388850 437 vklalDVEIA--TYRKLLEG-EECRLNgEGVGQVNVSVVQ 473
Cdd:COG3206 359 -----EVEVAreLYESLLQRlEEARLA-EALTVGNVRVID 392
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
127-384 |
4.36e-06 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 49.68 E-value: 4.36e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 127 LLTPLNLQIDPAIQRVRAEEREQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQGTKTVRQnleplfeqyINNLRR 206
Cdd:TIGR02169 273 LLEELNKKIKDLGEEEQLRVKEKIGELEAEIASLERSIAEKERELEDAEERLAKLEAEIDKLLAE---------IEELER 343
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 207 QLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLralyD 286
Cdd:TIGR02169 344 EIEEERKRRDKLTEEYAELKEELEDLRAELEEVDKEFAETRDELKDYREKLEKLKREINELKRELDRLQEELQRL----S 419
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 287 AELSQMQTHISdtsvvlsmdnnrnlDLDSIIAEVKAQYEEIAQRSRAEaeswyQTKYEELQVTAGRHGDDLRNTKQEIAE 366
Cdd:TIGR02169 420 EELADLNAAIA--------------GIEAKINELEEEKEDKALEIKKQ-----EWKLEQLAADLSKYEQELYDLKEEYDR 480
|
250
....*....|....*...
gi 194388850 367 INRMIQRLRSEIDHVKKQ 384
Cdd:TIGR02169 481 VEKELSKLQRELAEAEAQ 498
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
140-427 |
6.73e-06 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 49.02 E-value: 6.73e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 140 QRVRAEEREQIKTLNNKFASFIDKVQFLEQQN-------KVLDTKWTLLQEQGTKTVRQNLE-----PLFEQYINNLRRQ 207
Cdd:pfam01576 425 ERQRAELAEKLSKLQSELESVSSLLNEAEGKNiklskdvSSLESQLQDTQELLQEETRQKLNlstrlRQLEDERNSLQEQ 504
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 208 LDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLtdeinflralyDA 287
Cdd:pfam01576 505 LEEEEEAKRNVERQLSTLQAQLSDMKKKLEEDAGTLEALEEGKKRLQRELEALTQQLEEKAAAYDKL-----------EK 573
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 288 ELSQMQTHISDTSVVLsmDNNRNL---------DLDSIIAEVK---AQYEEiaQRSRAEAESwyqTKYEELQVTAGRHGD 355
Cdd:pfam01576 574 TKNRLQQELDDLLVDL--DHQRQLvsnlekkqkKFDQMLAEEKaisARYAE--ERDRAEAEA---REKETRALSLARALE 646
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 194388850 356 DLRNTKQEIAEINRMIQ----RLRSEIDHVKKQCASLQAAIADAEQrgemALKDAKNKLEGLEDALQKAKQDLARL 427
Cdd:pfam01576 647 EALEAKEELERTNKQLRaemeDLVSSKDDVGKNVHELERSKRALEQ----QVEEMKTQLEELEDELQATEDAKLRL 718
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
191-456 |
7.24e-06 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 48.91 E-value: 7.24e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 191 QNLEPLFEQyINNLRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEdEVNKRTAAENEfvtLKKDVDaAYMNKVELQAK 270
Cdd:PRK03918 231 KELEELKEE-IEELEKELESLEGSKRKLEEKIRELEERIEELKKEIE-ELEEKVKELKE---LKEKAE-EYIKLSEFYEE 304
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 271 ADTLTDEINFLRALYDAELSQMQTHISDtsvvLSMDNNRNLDLDSIIAEVKAQYEEIAQRSRAeaeswyqtkYEELQVTA 350
Cdd:PRK03918 305 YLDELREIEKRLSRLEEEINGIEERIKE----LEEKEERLEELKKKLKELEKRLEELEERHEL---------YEEAKAKK 371
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 351 GRhgddLRNTKQEIA-----EINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGE------MALKDAKNKL--------- 410
Cdd:PRK03918 372 EE----LERLKKRLTgltpeKLEKELEELEKAKEEIEEEISKITARIGELKKEIKelkkaiEELKKAKGKCpvcgrelte 447
|
250 260 270 280
....*....|....*....|....*....|....*....|....*.
gi 194388850 411 EGLEDALQKAKQDLARLLKEYQELMNVKLALDVEIATYRKLLEGEE 456
Cdd:PRK03918 448 EHRKELLEEYTAELKRIEKELKEIEEKERKLRKELRELEKVLKKES 493
|
|
| CwlO1 |
COG3883 |
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function ... |
201-420 |
2.94e-05 |
|
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function unknown];
Pssm-ID: 443091 [Multi-domain] Cd Length: 379 Bit Score: 46.36 E-value: 2.94e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 201 INNLRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDaaymnkvELQAKADTLTDEI-N 279
Cdd:COG3883 18 IQAKQKELSELQAELEAAQAELDALQAELEELNEEYNELQAELEALQAEIDKLQAEIA-------EAEAEIEERREELgE 90
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 280 FLRALYDAELSqmqthISDTSVVLSMDN-----NRNLDLDSIIAEVKAQYEEI--AQRSRAEAESWYQTKYEELQVTAGR 352
Cdd:COG3883 91 RARALYRSGGS-----VSYLDVLLGSESfsdflDRLSALSKIADADADLLEELkaDKAELEAKKAELEAKLAELEALKAE 165
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....*...
gi 194388850 353 HGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKA 420
Cdd:COG3883 166 LEAAKAELEAQQAEQEALLAQLSAEEAAAEAQLAELEAELAAAEAAAAAAAAAAAAAAAAAAAAAAAA 233
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
356-437 |
5.44e-05 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 45.53 E-value: 5.44e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 356 DLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQR---GEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQ 432
Cdd:COG4942 28 ELEQLQQEIAELEKELAALKKEEKALLKQLAALERRIAALARRiraLEQELAALEAELAELEKEIAELRAELEAQKEELA 107
|
....*
gi 194388850 433 ELMNV 437
Cdd:COG4942 108 ELLRA 112
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
355-434 |
6.59e-05 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 45.14 E-value: 6.59e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 355 DDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRgemaLKDAKNKLEGLEDALQKAKQDLARLLKEYQEL 434
Cdd:COG4942 20 DAAAEAEAELEQLQQEIAELEKELAALKKEEKALLKQLAALERR----IAALARRIRALEQELAALEAELAELEKEIAEL 95
|
|
| 46 |
PHA02562 |
endonuclease subunit; Provisional |
162-430 |
6.64e-05 |
|
endonuclease subunit; Provisional
Pssm-ID: 222878 [Multi-domain] Cd Length: 562 Bit Score: 45.78 E-value: 6.64e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 162 DKVQFLEQQNKVLDTKWTLLQEQgTKTvrqnleplFEQYINNLRRQLDSIVgergrldSELRNMQDLVEDLKNKYEDEVN 241
Cdd:PHA02562 174 DKIRELNQQIQTLDMKIDHIQQQ-IKT--------YNKNIEEQRKKNGENI-------ARKQNKYDELVEEAKTIKAEIE 237
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 242 KRTAAENEFVTLKKDVDAAY----MNKVELQAKADTLTDEINFLRAlYDAELSQMQThISDTsvvlsmdnnrnldlDSII 317
Cdd:PHA02562 238 ELTDELLNLVMDIEDPSAALnklnTAAAKIKSKIEQFQKVIKMYEK-GGVCPTCTQQ-ISEG--------------PDRI 301
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 318 AEVKAQYEEIAQRSRAEaeswyQTKYEELQVTAGRHgDDLRNTKQE----IAEINRMIQRLRSEIDHVKKQCASLQAAIa 393
Cdd:PHA02562 302 TKIKDKLKELQHSLEKL-----DTAIDELEEIMDEF-NEQSKKLLElknkISTNKQSLITLVDKAKKVKAAIEELQAEF- 374
|
250 260 270
....*....|....*....|....*....|....*..
gi 194388850 394 daeqrgemalKDAKNKLEGLEDALQKAKQDLARLLKE 430
Cdd:PHA02562 375 ----------VDNAEELAKLQDELDKIVKTKSELVKE 401
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
214-434 |
8.13e-05 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 45.14 E-value: 8.13e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 214 ERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLRalydAELSQMQ 293
Cdd:COG4942 28 ELEQLQQEIAELEKELAALKKEEKALLKQLAALERRIAALARRIRALEQELAALEAELAELEKEIAELR----AELEAQK 103
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 294 THISDTSVVLSMDNNRNldldsiIAEVKAQYEEIAQRSRAEAesWYQTKYEELQvtagRHGDDLRNTKQEIAEINRMIQR 373
Cdd:COG4942 104 EELAELLRALYRLGRQP------PLALLLSPEDFLDAVRRLQ--YLKYLAPARR----EQAEELRADLAELAALRAELEA 171
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|.
gi 194388850 374 LRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQEL 434
Cdd:COG4942 172 ERAELEALLAELEEERAALEALKAERQKLLARLEKELAELAAELAELQQEAEELEALIARL 232
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
312-450 |
9.44e-05 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 45.29 E-value: 9.44e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 312 DLDSIIAEVKAQYEEIAQRsRAEAEswyqTKYEELQVT-AGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQA 390
Cdd:COG4913 299 ELRAELARLEAELERLEAR-LDALR----EELDELEAQiRGNGGDRLEQLEREIERLERELEERERRRARLEALLAALGL 373
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 391 AIADAEQRGEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQELMNVKLALDVEIATYRK 450
Cdd:COG4913 374 PLPASAEEFAALRAEAAALLEALEEELEALEEALAEAEAALRDLRRELRELEAEIASLER 433
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
355-461 |
1.17e-04 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 45.29 E-value: 1.17e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 355 DDLRNTKQEIAEINRMIQRLRSEIDHVKKQC----------------ASLQAAIADAEQRGEmALKDAKNKLEGLEDALQ 418
Cdd:COG4913 617 AELAELEEELAEAEERLEALEAELDALQERRealqrlaeyswdeidvASAEREIAELEAELE-RLDASSDDLAALEEQLE 695
|
90 100 110 120
....*....|....*....|....*....|....*....|...
gi 194388850 419 KAKQDLARLLKEYQELMNVKLALDVEIATYRKLLEGEECRLNG 461
Cdd:COG4913 696 ELEAELEELEEELDELKGEIGRLEKELEQAEEELDELQDRLEA 738
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
205-436 |
1.32e-04 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 44.91 E-value: 1.32e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 205 RRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAymnkvELQAKADTLTDEInflral 284
Cdd:COG4913 609 RAKLAALEAELAELEEELAEAEERLEALEAELDALQERREALQRLAEYSWDEIDVA-----SAEREIAELEAEL------ 677
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 285 ydaelsqmqthisdtsvvlsmdnnRNLDLDS-IIAEVKAQYEEiAQRSRAEAESwyqtKYEELQVTAGRHgddlrntKQE 363
Cdd:COG4913 678 ------------------------ERLDASSdDLAALEEQLEE-LEAELEELEE----ELDELKGEIGRL-------EKE 721
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 194388850 364 IAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQELMN 436
Cdd:COG4913 722 LEQAEEELDELQDRLEAAEDLARLELRALLEERFAAALGDAVERELRENLEERIDALRARLNRAEEELERAMR 794
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
312-453 |
1.53e-04 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 43.37 E-value: 1.53e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 312 DLDSIIAEVKAQYEEI-AQRSRAEAE-SWYQTKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVK--KQCAS 387
Cdd:COG1579 14 ELDSELDRLEHRLKELpAELAELEDElAALEARLEAAKTELEDLEKEIKRLELEIEEVEARIKKYEEQLGNVRnnKEYEA 93
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 194388850 388 LQAAIADAEQR---GEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQELmnvKLALDVEIATYRKLLE 453
Cdd:COG1579 94 LQKEIESLKRRisdLEDEILELMERIEELEEELAELEAELAELEAELEEK---KAELDEELAELEAELE 159
|
|
| CwlO1 |
COG3883 |
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function ... |
246-450 |
3.21e-04 |
|
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function unknown];
Pssm-ID: 443091 [Multi-domain] Cd Length: 379 Bit Score: 43.28 E-value: 3.21e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 246 AENEFVTLKKDVDAAYMNKVELQAKADTLTDEINFLRALYD---AELSQMQTHISDTsvvlsmdnnrNLDLDSIIAEVKA 322
Cdd:COG3883 14 ADPQIQAKQKELSELQAELEAAQAELDALQAELEELNEEYNelqAELEALQAEIDKL----------QAEIAEAEAEIEE 83
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 323 QYEEIAQRSRAEAESWYQTKYEELQVTAGRHGDDLRNtkqeIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEmA 402
Cdd:COG3883 84 RREELGERARALYRSGGSVSYLDVLLGSESFSDFLDR----LSALSKIADADADLLEELKADKAELEAKKAELEAKLA-E 158
|
170 180 190 200 210
....*....|....*....|....*....|....*....|....*....|..
gi 194388850 403 LKDAKNKLEGLEDALQKAKQD----LARLLKEYQELMNVKLALDVEIATYRK 450
Cdd:COG3883 159 LEALKAELEAAKAELEAQQAEqealLAQLSAEEAAAEAQLAELEAELAAAEA 210
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
317-447 |
3.27e-04 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 43.77 E-value: 3.27e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 317 IAEVKAQYEEIAQRSRAEAEswyqtKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAE 396
Cdd:COG1196 262 LAELEAELEELRLELEELEL-----ELEEAQAEEYELLAELARLEQDIARLEERRRELEERLEELEEELAELEEELEELE 336
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....
gi 194388850 397 QR---GEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQELMNVKLALDVEIAT 447
Cdd:COG1196 337 EEleeLEEELEEAEEELEEAEAELAEAEEALLEAEAELAEAEEELEELAEELLE 390
|
|
| SCP-1 |
pfam05483 |
Synaptonemal complex protein 1 (SCP-1); Synaptonemal complex protein 1 (SCP-1) is the major ... |
139-438 |
5.88e-04 |
|
Synaptonemal complex protein 1 (SCP-1); Synaptonemal complex protein 1 (SCP-1) is the major component of the transverse filaments of the synaptonemal complex. Synaptonemal complexes are structures that are formed between homologous chromosomes during meiotic prophase.
Pssm-ID: 114219 [Multi-domain] Cd Length: 787 Bit Score: 42.79 E-value: 5.88e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 139 IQRVRAEEREQIKTLNNKFASfidkvqfLEQQNKVLDTKWTLLQE-----------QGTKTVRQNLEPLFEQYINNLRRQ 207
Cdd:pfam05483 386 LQKKSSELEEMTKFKNNKEVE-------LEELKKILAEDEKLLDEkkqfekiaeelKGKEQELIFLLQAREKEIHDLEIQ 458
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 208 LDSIVGERGRLDSElrnmqdlVEDLKNKYEDEVNKRTAaenefvtLKKDVDAAYMNKVELQAKADTLTDEINFLRALYDA 287
Cdd:pfam05483 459 LTAIKTSEEHYLKE-------VEDLKTELEKEKLKNIE-------LTAHCDKLLLENKELTQEASDMTLELKKHQEDIIN 524
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 288 ELSQMQTHISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEIAQRSRAEAESWYQTKYEELQVTAGRHGDDLR--NTKQEIA 365
Cdd:pfam05483 525 CKKQEERMLKQIENLEEKEMNLRDELESVREEFIQKGDEVKCKLDKSEENARSIEYEVLKKEKQMKILENKcnNLKKQIE 604
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 194388850 366 EINRMIQRLRSEIDHVKKQcaslqaaiADAEQRGEMALKDAKNKLEgLEdaLQKAKQDLARLLKEYQELMNVK 438
Cdd:pfam05483 605 NKNKNIEELHQENKALKKK--------GSAENKQLNAYEIKVNKLE-LE--LASAKQKFEEIIDNYQKEIEDK 666
|
|
| 235kDa-fam |
TIGR01612 |
reticulocyte binding/rhoptry protein; This model represents a group of paralogous families in ... |
145-419 |
6.99e-04 |
|
reticulocyte binding/rhoptry protein; This model represents a group of paralogous families in plasmodium species alternately annotated as reticulocyte binding protein, 235-kDa family protein and rhoptry protein. Rhoptry protein is localized on the cell surface and is extremely large (although apparently lacking in repeat structure) and is important for the process of invasion of the RBCs by the parasite. These proteins are found in P. falciparum, P. vivax and P. yoelii.
Pssm-ID: 130673 [Multi-domain] Cd Length: 2757 Bit Score: 42.73 E-value: 6.99e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 145 EEREQIKTLNNKFASFIDKVQFLE-------------QQNKVLDTkWTLLQEQGTKTVRQNLEPLFEQYiNNLRRQLDSI 211
Cdd:TIGR01612 693 EDKAKLDDLKSKIDKEYDKIQNMEtatvelhlsnienKKNELLDI-IVEIKKHIHGEINKDLNKILEDF-KNKEKELSNK 770
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 212 VGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEfvtLKKDVDAAYMNKVELQAKADTLTDEINFLRALYDAELSQ 291
Cdd:TIGR01612 771 INDYAKEKDELNKYKSKISEIKNHYNDQINIDNIKDED---AKQNYDKSKEYIKTISIKEDEIFKIINEMKFMKDDFLNK 847
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 292 MQTHIsdtsvvlSMDNNRNLDLDSiiaeVKAQYEEIAQRSRAEAESWYQTKYEelqvtagrhgDDLRNTKQEIAEINRMI 371
Cdd:TIGR01612 848 VDKFI-------NFENNCKEKIDS----EHEQFAELTNKIKAEISDDKLNDYE----------KKFNDSKSLINEINKSI 906
|
250 260 270 280
....*....|....*....|....*....|....*....|....*...
gi 194388850 372 QRLRSEIDHVKKQCASLQAAIADAEqrgemALKDAKNKLEGLEDALQK 419
Cdd:TIGR01612 907 EEEYQNINTLKKVDEYIKICENTKE-----SIEKFHNKQNILKEILNK 949
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
201-457 |
7.01e-04 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 42.72 E-value: 7.01e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 201 INNLRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTaaenEFVTLKKDVDAAYMNKVELQAKADTLTDEINF 280
Cdd:PRK02224 208 LNGLESELAELDEEIERYEEQREQARETRDEADEVLEEHEERRE----ELETLEAEIEDLRETIAETEREREELAEEVRD 283
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 281 LRALYDaELSQMQTHISDTSVVLSMDNN----RNLDLDSIIAEVKAQYEEI---AQRSRAEAESW------YQTKYEELQ 347
Cdd:PRK02224 284 LRERLE-ELEEERDDLLAEAGLDDADAEaveaRREELEDRDEELRDRLEECrvaAQAHNEEAESLredaddLEERAEELR 362
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 348 VTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDhvkkqcaSLQAAIADAeqrgEMALKDAKNKLEGLEDALQKAKQDLARL 427
Cdd:PRK02224 363 EEAAELESELEEAREAVEDRREEIEELEEEIE-------ELRERFGDA----PVDLGNAEDFLEELREERDELREREAEL 431
|
250 260 270
....*....|....*....|....*....|
gi 194388850 428 LKEYQELMNVklaldveIATYRKLLEGEEC 457
Cdd:PRK02224 432 EATLRTARER-------VEEAEALLEAGKC 454
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
319-456 |
7.64e-04 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 42.62 E-value: 7.64e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 319 EVKAQYEEIAQRSRAEAESWYQTKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQR 398
Cdd:COG1196 217 ELKEELKELEAELLLLKLRELEAELEELEAELEELEAELEELEAELAELEAELEELRLELEELELELEEAQAEEYELLAE 296
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*...
gi 194388850 399 gemaLKDAKNKLEGLEDALQKAKQDLARLLKEYQELMNVKLALDVEIATYRKLLEGEE 456
Cdd:COG1196 297 ----LARLEQDIARLEERRRELEERLEELEEELAELEEELEELEEELEELEEELEEAE 350
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
312-459 |
9.28e-04 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 42.21 E-value: 9.28e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 312 DLDSIIAEVKAQYEEIAQRSRA----EAESWYQTKYEELQVTAGRHGD---DLRNTKQEIAEINRMIQRLRSEIDHVKKQ 384
Cdd:COG4913 628 EAEERLEALEAELDALQERREAlqrlAEYSWDEIDVASAEREIAELEAeleRLDASSDDLAALEEQLEELEAELEELEEE 707
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 385 CASLQAAIADAEQRgemaLKDAKNKLEGLEDALQKA-----KQDLARLLKEYQELM------NVKLALDVEIATYRKLLE 453
Cdd:COG4913 708 LDELKGEIGRLEKE----LEQAEEELDELQDRLEAAedlarLELRALLEERFAAALgdaverELRENLEERIDALRARLN 783
|
....*.
gi 194388850 454 GEECRL 459
Cdd:COG4913 784 RAEEEL 789
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
317-450 |
9.42e-04 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 42.06 E-value: 9.42e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 317 IAEVKAQYEEIAQRsRAEAESWYQTKYEELQVTAGRHgdDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAE 396
Cdd:COG4717 390 ALEQAEEYQELKEE-LEELEEQLEELLGELEELLEAL--DEEELEEELEELEEELEELEEELEELREELAELEAELEQLE 466
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....
gi 194388850 397 QRGEmalkdaknkLEGLEDALQKAKQDLARLLKEYQELMNVKLALDVEIATYRK 450
Cdd:COG4717 467 EDGE---------LAELLQELEELKAELRELAEEWAALKLALELLEEAREEYRE 511
|
|
| sbcc |
TIGR00618 |
exonuclease SbcC; All proteins in this family for which functions are known are part of an ... |
145-458 |
9.57e-04 |
|
exonuclease SbcC; All proteins in this family for which functions are known are part of an exonuclease complex with sbcD homologs. This complex is involved in the initiation of recombination to regulate the levels of palindromic sequences in DNA. This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University). [DNA metabolism, DNA replication, recombination, and repair]
Pssm-ID: 129705 [Multi-domain] Cd Length: 1042 Bit Score: 42.26 E-value: 9.57e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 145 EEREQIKTLNNKFASFIDKVQFLEQQnkvldtkWTLLQEQGTKTvrQNLEPLFEQYINNLRRQLDSIVGERGRLDSELR- 223
Cdd:TIGR00618 553 SERKQRASLKEQMQEIQQSFSILTQC-------DNRSKEDIPNL--QNITVRLQDLTEKLSEAEDMLACEQHALLRKLQp 623
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 224 --NMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDvdaaymnKVELQAKADTLTDEINFLRALydAELSQMQTHISDTSV 301
Cdd:TIGR00618 624 eqDLQDVRLHLQQCSQELALKLTALHALQLTLTQE-------RVREHALSIRVLPKELLASRQ--LALQKMQSEKEQLTY 694
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 302 VLSMDNNRNLDLDSI---IAEVKAQYEEIAQRSRA---------EAESWYQTKYEELQVTAGRHG--DDLRNTKQEIAEI 367
Cdd:TIGR00618 695 WKEMLAQCQTLLRELethIEEYDREFNEIENASSSlgsdlaareDALNQSLKELMHQARTVLKARteAHFNNNEEVTAAL 774
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 368 NRM--IQRLRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKAKQDLARLLKEyqelmnvKLALDVEI 445
Cdd:TIGR00618 775 QTGaeLSHLAAEIQFFNRLREEDTHLLKTLEAEIGQEIPSDEDILNLQCETLVQEEEQFLSRLEE-------KSATLGEI 847
|
330
....*....|...
gi 194388850 446 AtyRKLLEGEECR 458
Cdd:TIGR00618 848 T--HQLLKYEECS 858
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
119-298 |
1.24e-03 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 41.82 E-value: 1.24e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 119 QEVTVNQSLLTPLNLQIDpaiQRVRAEEREQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQGTKTVRQNLEPLfE 198
Cdd:COG4913 269 ERLAELEYLRAALRLWFA---QRRLELLEAELEELRAELARLEAELERLEARLDALREELDELEAQIRGNGGDRLEQL-E 344
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 199 QYINNLRRQLDSIVGERGRLDSELRNMQ----DLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTL 274
Cdd:COG4913 345 REIERLERELEERERRRARLEALLAALGlplpASAEEFAALRAEAAALLEALEEELEALEEALAEAEAALRDLRRELREL 424
|
170 180
....*....|....*....|....*..
gi 194388850 275 TDEINFLRA---LYDAELSQMQTHISD 298
Cdd:COG4913 425 EAEIASLERrksNIPARLLALRDALAE 451
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
188-434 |
1.39e-03 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 40.29 E-value: 1.39e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 188 TVRQNLEPLFEqyINNLRRQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDaaymnkvEL 267
Cdd:COG1579 1 AMPEDLRALLD--LQELDSELDRLEHRLKELPAELAELEDELAALEARLEAAKTELEDLEKEIKRLELEIE-------EV 71
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 268 QAKADTLTDEINFLRalydaelsqmqthisdtsvvlsmdNNRnlDLDSIIAEVkaqyeEIAQRSRAEAEswyqtkyeelq 347
Cdd:COG1579 72 EARIKKYEEQLGNVR------------------------NNK--EYEALQKEI-----ESLKRRISDLE----------- 109
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 348 vtagrhgddlrntkQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKAKQDL-AR 426
Cdd:COG1579 110 --------------DEILELMERIEELEEELAELEAELAELEAELEEKKAELDEELAELEAELEELEAEREELAAKIpPE 175
|
....*...
gi 194388850 427 LLKEYQEL 434
Cdd:COG1579 176 LLALYERI 183
|
|
| DUF1351 |
pfam07083 |
Protein of unknown function (DUF1351); This family consists of several bacterial and phage ... |
310-445 |
1.53e-03 |
|
Protein of unknown function (DUF1351); This family consists of several bacterial and phage proteins of around 230 residues in length. The function of this family is unknown.
Pssm-ID: 429283 [Multi-domain] Cd Length: 210 Bit Score: 40.05 E-value: 1.53e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 310 NLDLDSIIAEVKAQYEEIAQRSRAEAEswyqtKYEELQVTAgrhgDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCAslq 389
Cdd:pfam07083 1 ELSVTQKPAAISFNFEELETYVDGIVA-----KYEGLVVTE----DTVKEAKKERAELNKIAKALDDKRKEVKKQYS--- 68
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....
gi 194388850 390 AAIADAEQRG---EMALKDAKNKL----EGLEDALQKAKQDLARLLK-EYQELMNVKLAlDVEI 445
Cdd:pfam07083 69 EPYDEFEAKIkelVAKIKEAIDPIdeqiKAFEEKEKDAKRQLVKALIsELAEEYGVPLE-EIEI 131
|
|
| 235kDa-fam |
TIGR01612 |
reticulocyte binding/rhoptry protein; This model represents a group of paralogous families in ... |
139-429 |
1.77e-03 |
|
reticulocyte binding/rhoptry protein; This model represents a group of paralogous families in plasmodium species alternately annotated as reticulocyte binding protein, 235-kDa family protein and rhoptry protein. Rhoptry protein is localized on the cell surface and is extremely large (although apparently lacking in repeat structure) and is important for the process of invasion of the RBCs by the parasite. These proteins are found in P. falciparum, P. vivax and P. yoelii.
Pssm-ID: 130673 [Multi-domain] Cd Length: 2757 Bit Score: 41.58 E-value: 1.77e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 139 IQRVRAEEREQIKTLNNK---FASFIDKVQFLEQQNKVLD----------TKWTLLQEQGTKTVRQNLEPLFEQYINNLR 205
Cdd:TIGR01612 598 INKLKLELKEKIKNISDKneyIKKAIDLKKIIENNNAYIDelakispyqvPEHLKNKDKIYSTIKSELSKIYEDDIDALY 677
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 206 RQLDSIVGErgrldSELRNMQD--LVEDLKNKYEDEVNKRTAAENEfvTLKKDVDAAYMNKVELQAKA--------DTLT 275
Cdd:TIGR01612 678 NELSSIVKE-----NAIDNTEDkaKLDDLKSKIDKEYDKIQNMETA--TVELHLSNIENKKNELLDIIveikkhihGEIN 750
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 276 DEINFLRALYDAELSQMQTHISDTSvvlsmdnNRNLDLD---SIIAEVKAQYEE---IAQRSRAEAESWYQTKYEELQVT 349
Cdd:TIGR01612 751 KDLNKILEDFKNKEKELSNKINDYA-------KEKDELNkykSKISEIKNHYNDqinIDNIKDEDAKQNYDKSKEYIKTI 823
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 350 AGRHGD------DLRNTKQEI-AEINRMI-------QRLRSEIDHVKKQCASLQAAIADAEQRG-EMALKDAKNKLEGLE 414
Cdd:TIGR01612 824 SIKEDEifkiinEMKFMKDDFlNKVDKFInfennckEKIDSEHEQFAELTNKIKAEISDDKLNDyEKKFNDSKSLINEIN 903
|
330
....*....|....*
gi 194388850 415 DALQKAKQDLARLLK 429
Cdd:TIGR01612 904 KSIEEEYQNINTLKK 918
|
|
| ClyA_Cry6Aa-like |
cd22656 |
Bacillus thuringiensis crystal 6Aa (Cry6Aa) toxin, and similar proteins; This model includes ... |
216-433 |
2.10e-03 |
|
Bacillus thuringiensis crystal 6Aa (Cry6Aa) toxin, and similar proteins; This model includes pesticidal Cry6Aa toxin from Bacillus thuringiensis, one of the many parasporal crystal (Cry) toxins produced during the sporulation phase of growth. Many of these proteins are toxic to numerous insect species and have been effectively used as proteinaceous insecticides to directly kill insect pests; some have been used to control insect growth on transgenic agricultural plants. Cry6Aa exists as a protoxin, which is activated by cleavage using trypsin. Structure studies for Cry6Aa support a mechanism of action by pore formation, similar to cytolysin A (ClyA)-type alpha pore-forming toxins (alpha-PFTs) such as HblB, and bioassay and mutation studies show that Cry6Aa is an active pore-forming toxin. Cry6Aa shows atypical features compared to other members of alpha-PFTs, including internal repeat sequences and small loop regions within major alpha helices.
Pssm-ID: 439154 [Multi-domain] Cd Length: 309 Bit Score: 40.43 E-value: 2.10e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 216 GRLDSELRNMQDLVEDLKNKYEDEVNKRTAAE-NEFV-TLKKDVDaaymnkvELQAKADTLTDEIN-FLRAL--YDAELS 290
Cdd:cd22656 87 GTIDSYYAEILELIDDLADATDDEELEEAKKTiKALLdDLLKEAK-------KYQDKAAKVVDKLTdFENQTekDQTALE 159
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 291 QMQTHISDtsvVLSMDNNRNL--DLDSIIAEVKAQYEEIAQRSRA---EAESWYQTKYEELQV------TAGRHGDDLRN 359
Cdd:cd22656 160 TLEKALKD---LLTDEGGAIArkEIKDLQKELEKLNEEYAAKLKAkidELKALIADDEAKLAAalrliaDLTAADTDLDN 236
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 194388850 360 TKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQrgemALKDAKNKLEGLEDALQKAKqDLARLLKEYQE 433
Cdd:cd22656 237 LLALIGPAIPALEKLQGAWQAIATDLDSLKDLLEDDIS----KIPAAILAKLELEKAIEKWN-ELAEKADKFRQ 305
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
148-433 |
2.94e-03 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 40.39 E-value: 2.94e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 148 EQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQgTKTVRQNLEPLfEQYINNLRRQLDSIVGERGRLDSELRNMQD 227
Cdd:TIGR04523 75 NKIKILEQQIKDLNDKLKKNKDKINKLNSDLSKINSE-IKNDKEQKNKL-EVELNKLEKQKKENKKNIDKFLTEIKKKEK 152
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 228 LVEDLKNKYEDEVNKRTAAENEFVTLKKDvdaaymnKVELQAKADTLTDEINFLRALydaeLSQMQTHISdtsvvlsmdn 307
Cdd:TIGR04523 153 ELEKLNNKYNDLKKQKEELENELNLLEKE-------KLNIQKNIDKIKNKLLKLELL----LSNLKKKIQ---------- 211
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 308 nRNLDLDSIIAEVKAQYEEIaqrsraeaeswyQTKYEELQvtagrhgDDLRNTKQEIAEINRMIQRLRSEIDHVKKQcas 387
Cdd:TIGR04523 212 -KNKSLESQISELKKQNNQL------------KDNIEKKQ-------QEINEKTTEISNTQTQLNQLKDEQNKIKKQ--- 268
|
250 260 270 280
....*....|....*....|....*....|....*....|....*.
gi 194388850 388 LQAAIADAEQrgemalkdAKNKLEGLEDALQKAKQDLARLLKEYQE 433
Cdd:TIGR04523 269 LSEKQKELEQ--------NNKKIKELEKQLNQLKSEISDLNNQKEQ 306
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
355-468 |
2.97e-03 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 40.44 E-value: 2.97e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 355 DDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQ-------RGEMALKD---AKNKLEGLEDALQKAKQDL 424
Cdd:TIGR02169 674 AELQRLRERLEGLKRELSSLQSELRRIENRLDELSQELSDASRkigeiekEIEQLEQEeekLKERLEELEEDLSSLEQEI 753
|
90 100 110 120
....*....|....*....|....*....|....*....|....
gi 194388850 425 ARLLKEYQELMNVKLALDVEIATYRKLLEGEECRLNGEGVGQVN 468
Cdd:TIGR02169 754 ENVKSELKELEARIEELEEDLHKLEEALNDLEARLSHSRIPEIQ 797
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
136-439 |
6.45e-03 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 39.25 E-value: 6.45e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 136 DPAIQRVRAEEREQIKTLNNKFASFIDKVQFLEQQNKVLDTKWTLLQEQGTKTVRQNLEplFEQYINNLRRQLDSIVGER 215
Cdd:PRK02224 323 DEELRDRLEECRVAAQAHNEEAESLREDADDLEERAEELREEAAELESELEEAREAVED--RREEIEELEEEIEELRERF 400
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 216 GRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYM-----------NKVELQAKADTLTDEINFLRAL 284
Cdd:PRK02224 401 GDAPVDLGNAEDFLEELREERDELREREAELEATLRTARERVEEAEAlleagkcpecgQPVEGSPHVETIEEDRERVEEL 480
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 285 yDAELSQMQTHISDTSVVL--------------SMDNNRNlDLDSIIAEVKAQYEE---IAQRSRAEAESwYQTKYEELQ 347
Cdd:PRK02224 481 -EAELEDLEEEVEEVEERLeraedlveaedrieRLEERRE-DLEELIAERRETIEEkreRAEELRERAAE-LEAEAEEKR 557
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 348 VTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKqCASLQAAIADAEQR-----------GEM---------ALKDAK 407
Cdd:PRK02224 558 EAAAEAEEEAEEAREEVAELNSKLAELKERIESLER-IRTLLAAIADAEDEierlrekrealAELnderrerlaEKRERK 636
|
330 340 350
....*....|....*....|....*....|....
gi 194388850 408 NKLEGL--EDALQKAKQDLARlLKEYQELMNVKL 439
Cdd:PRK02224 637 RELEAEfdEARIEEAREDKER-AEEYLEQVEEKL 669
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
364-459 |
6.86e-03 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 39.28 E-value: 6.86e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 364 IAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEmALKDAKNKLEGLeDALQKAKQDLA--RLLKEYQELMNVKLAL 441
Cdd:TIGR02169 165 VAEFDRKKEKALEELEEVEENIERLDLIIDEKRQQLE-RLRREREKAERY-QALLKEKREYEgyELLKEKEALERQKEAI 242
|
90
....*....|....*...
gi 194388850 442 DVEIATYRKLLEGEECRL 459
Cdd:TIGR02169 243 ERQLASLEEELEKLTEEI 260
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
317-427 |
6.93e-03 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 39.51 E-value: 6.93e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 317 IAEVKAQYEEIaQRSRAEAESWY-QTKYEELQvtagrhgDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADA 395
Cdd:COG4913 264 YAAARERLAEL-EYLRAALRLWFaQRRLELLE-------AELEELRAELARLEAELERLEARLDALREELDELEAQIRGN 335
|
90 100 110
....*....|....*....|....*....|..
gi 194388850 396 EQRgemALKDAKNKLEGLEDALQKAKQDLARL 427
Cdd:COG4913 336 GGD---RLEQLEREIERLERELEERERRRARL 364
|
|
| YqiK |
COG2268 |
Uncharacterized membrane protein YqiK, contains Band7/PHB/SPFH domain [Function unknown]; |
317-428 |
7.47e-03 |
|
Uncharacterized membrane protein YqiK, contains Band7/PHB/SPFH domain [Function unknown];
Pssm-ID: 441869 [Multi-domain] Cd Length: 439 Bit Score: 39.09 E-value: 7.47e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 317 IAEVKAQYEEIAQRSRAEAESWYQTKYEELQVTAGRHGDDLRNTKQ-EIAEINRMIQRLRSEIDHVKKQCASLQAAIADA 395
Cdd:COG2268 246 LAKKKAEERREAETARAEAEAAYEIAEANAEREVQRQLEIAEREREiELQEKEAEREEAELEADVRKPAEAEKQAAEAEA 325
|
90 100 110
....*....|....*....|....*....|....*.
gi 194388850 396 EQRGEMALKDAKNKLEGLE---DALQKAKQDLARLL 428
Cdd:COG2268 326 EAEAEAIRAKGLAEAEGKRalaEAWNKLGDAAILLM 361
|
|
| PRK12704 |
PRK12704 |
phosphodiesterase; Provisional |
318-433 |
8.33e-03 |
|
phosphodiesterase; Provisional
Pssm-ID: 237177 [Multi-domain] Cd Length: 520 Bit Score: 38.99 E-value: 8.33e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 318 AEVKAQYEEIAQRSRAEAE-----SWYQTKYEELQVTAGRHGDDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAI 392
Cdd:PRK12704 58 ALLEAKEEIHKLRNEFEKElrerrNELQKLEKRLLQKEENLDRKLELLEKREEELEKKEKELEQKQQELEKKEEELEELI 137
|
90 100 110 120
....*....|....*....|....*....|....*....|....*
gi 194388850 393 ADAEQRGE----MALKDAKNKLegLEDALQKAKQDLARLLKEYQE 433
Cdd:PRK12704 138 EEQLQELErisgLTAEEAKEIL--LEKVEEEARHEAAVLIKEIEE 180
|
|
| Spc7 |
smart00787 |
Spc7 kinetochore protein; This domain is found in cell division proteins which are required ... |
355-459 |
8.73e-03 |
|
Spc7 kinetochore protein; This domain is found in cell division proteins which are required for kinetochore-spindle association.
Pssm-ID: 197874 [Multi-domain] Cd Length: 312 Bit Score: 38.46 E-value: 8.73e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 355 DDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAEQRGEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQEL 434
Cdd:smart00787 158 EDYKLLMKELELLNSIKPKLRDRKDALEEELRQLKQLEDELEDCDPTELDRAKEKLKKLLQEIMIKVKKLEELEEELQEL 237
|
90 100 110
....*....|....*....|....*....|..
gi 194388850 435 -------MNVKLALDVEIATYRKLLegEECRL 459
Cdd:smart00787 238 eskiedlTNKKSELNTEIAEAEKKL--EQCRG 267
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
206-434 |
9.25e-03 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 38.90 E-value: 9.25e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 206 RQLDSIVGERGRLDSELRNMQDLVEDLKNKYEDEVNKRTAAENEFVTLKKDVDAAYMNKVELQAKADTLTDEI------- 278
Cdd:TIGR02169 716 RKIGEIEKEIEQLEQEEEKLKERLEELEEDLSSLEQEIENVKSELKELEARIEELEEDLHKLEEALNDLEARLshsripe 795
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 279 --NFLRALyDAELSQMQTHISDTSVVLSMDNNRNLDLDSIIAEVKAQYEEIAQR--SRAEAESWYQTKYEELQvtagrhg 354
Cdd:TIGR02169 796 iqAELSKL-EEEVSRIEARLREIEQKLNRLTLEKEYLEKEIQELQEQRIDLKEQikSIEKEIENLNGKKEELE------- 867
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 194388850 355 DDLRNTKQEIAEINRMIQRLRSEIDHVKKQCASLQAAIADAeqrgEMALKDAKNKLEGLEDALQKAKQDLARLLKEYQEL 434
Cdd:TIGR02169 868 EELEELEAALRDLESRLGDLKKERDELEAQLRELERKIEEL----EAQIEKKRKRLSELKAKLEALEEELSEIEDPKGED 943
|
|
| |