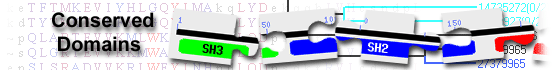|
Name |
Accession |
Description |
Interval |
E-value |
| Filament |
pfam00038 |
Intermediate filament protein; |
17-274 |
2.44e-72 |
|
Intermediate filament protein;
Pssm-ID: 459643 [Multi-domain] Cd Length: 313 Bit Score: 234.04 E-value: 2.44e-72
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARLKDL-EALLNSKEAALSTALSEKRTLEGELHDLRGQVAKL--------------EAALGEAKKQLQDEMLRRVDA 81
Cdd:pfam00038 43 AEPSRLYSLyEKEIEDLRRQLDTLTVERARLQLELDNLRLAAEDFrqkyedelnlrtsaENDLVGLRKDLDEATLARVDL 122
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 82 ENRLQTMKEELDFQKNIYSEELRETKRRH--ETRLVEIDNGKQREfesrLADALQELRAQHEDQVEQYKKELEKTYSAKL 159
Cdd:pfam00038 123 EAKIESLKEELAFLKKNHEEEVRELQAQVsdTQVNVEMDAARKLD----LTSALAEIRAQYEEIAAKNREEAEEWYQSKL 198
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 160 DNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRAR 239
Cdd:pfam00038 199 EELQQAAARNGDALRSAKEEITELRRTIQSLEIELQSLKKQKASLERQLAETEERYELQLADYQELISELEAELQETRQE 278
|
250 260 270
....*....|....*....|....*....|....*
gi 383792150 240 MQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEER 274
Cdd:pfam00038 279 MARQLREYQELLNVKLALDIEIATYRKLLEGEECR 313
|
|
| LTD |
pfam00932 |
Lamin Tail Domain; The lamin-tail domain (LTD), which has an immunoglobulin (Ig) fold, is ... |
322-429 |
2.85e-21 |
|
Lamin Tail Domain; The lamin-tail domain (LTD), which has an immunoglobulin (Ig) fold, is found in Nuclear Lamins, Chlo1887 from Chloroflexus, and several bacterial proteins where it occurs with membrane associated hydrolases of the metallo-beta-lactamase,synaptojanin, and calcineurin-like phosphoesterase superfamilies.
Pssm-ID: 460003 [Multi-domain] Cd Length: 108 Bit Score: 89.02 E-value: 2.85e-21
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 322 ARTSGRVAVEEVDEEG-----KFVRLRNKSNEDQSMGNWQIKRQNGDdpllTYRFPPKFTLKAGQVVTIWAAG----AGA 392
Cdd:pfam00932 1 SSATGDVVISEVVYDGsggndEFIELYNTGSKAVDLSGWKLQDASGG----TYTFPNGTTLAPGQTVVVWTGSgtnsATA 76
|
90 100 110
....*....|....*....|....*....|....*..
gi 383792150 393 THSPPTDLVWKAQNTWgcgnslrTALINSTGEEVAMR 429
Cdd:pfam00932 77 GYWGPSNAVWNNGGDA-------VALYDANGELVDSV 106
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
10-274 |
3.32e-16 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 82.29 E-value: 3.32e-16
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 10 KKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMK 89
Cdd:COG1196 243 ELEAELEELEAELEELEAELAELEAELEELRLELEELELELEEAQAEEYELLAELARLEQDIARLEERRRELEERLEELE 322
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 90 EELDfQKNIYSEELRETKRRHETRLVEIDNgKQREFESRLADALQELRAQHEDQVEQyKKELEKTYSAKLDNARQSAERN 169
Cdd:COG1196 323 EELA-ELEEELEELEEELEELEEELEEAEE-ELEEAEAELAEAEEALLEAEAELAEA-EEELEELAEELLEALRAAAELA 399
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 170 SNLVGAAhEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQE 249
Cdd:COG1196 400 AQLEELE-EAEEALLERLERLEEELEELEEALAELEEEEEEEEEALEEAAEEEAELEEEEEALLELLAELLEEAALLEAA 478
|
250 260
....*....|....*....|....*
gi 383792150 250 LLDIKLALDMEIHAYRKLLEGEEER 274
Cdd:COG1196 479 LAELLEELAEAAARLLLLLEAEADY 503
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
17-277 |
1.43e-12 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 70.74 E-value: 1.43e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARL-KDLEALLNSKEAALstALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELdfq 95
Cdd:COG1196 209 AEKAERyRELKEELKELEAEL--LLLKLRELEAELEELEAELEELEAELEELEAELAELEAELEELRLELEELELEL--- 283
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 96 kniysEELRETKRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEKtysAKLDNARQSAERNSNLVGA 175
Cdd:COG1196 284 -----EEAQAEEYELLAELARLEQDIARLEERRRELEERLEELEEELAELEEELEELE---EELEELEEELEEAEEELEE 355
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 176 AHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKL 255
Cdd:COG1196 356 AEAELAEAEEALLEAEAELAEAEEELEELAEELLEALRAAAELAAQLEELEEAEEALLERLERLEEELEELEEALAELEE 435
|
250 260
....*....|....*....|..
gi 383792150 256 ALDMEIHAYRKLLEGEEERLRL 277
Cdd:COG1196 436 EEEEEEEALEEAAEEEAELEEE 457
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
12-275 |
1.70e-12 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 70.47 E-value: 1.70e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 12 EGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRlqtmKEE 91
Cdd:TIGR02168 690 EEKIAELEKALAELRKELEELEEELEQLRKELEELSRQISALRKDLARLEAEVEQLEERIAQLSKELTELEAE----IEE 765
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 92 LDFQKNIYSEELRETKRRHETRLVEIDNGKQREFESRlaDALQELRAQHEDQveqykkelektySAKLDNARQSAERNSN 171
Cdd:TIGR02168 766 LEERLEEAEEELAEAEAEIEELEAQIEQLKEELKALR--EALDELRAELTLL------------NEEAANLRERLESLER 831
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 172 LVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLAR---ERDTSRRLLAEKEREMAEMRARMQQQLDEYQ 248
Cdd:TIGR02168 832 RIAATERRLEDLEEQIEELSEDIESLAAEIEELEELIEELESELEAllnERASLEEALALLRSELEELSEELRELESKRS 911
|
250 260
....*....|....*....|....*..
gi 383792150 249 ELLDIKLALDMEIHAYRKLLEGEEERL 275
Cdd:TIGR02168 912 ELRRELEELREKLAQLELRLEGLEVRI 938
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
49-335 |
2.28e-12 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 70.09 E-value: 2.28e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 49 ELHDLRGQVAKLEAALGEAKKQLQdemlrrvDAENRLQTMKEELdfqkniysEELRETKRRHETRLVEIDNGKQR----- 123
Cdd:TIGR02168 678 EIEELEEKIEELEEKIAELEKALA-------ELRKELEELEEEL--------EQLRKELEELSRQISALRKDLARleaev 742
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 124 EFESRLADALQELRAQHEDQVEQYKKELEKTYSAKLDNARQSAERNSnLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAA 203
Cdd:TIGR02168 743 EQLEERIAQLSKELTELEAEIEELEERLEEAEEELAEAEAEIEELEA-QIEQLKEELKALREALDELRAELTLLNEEAAN 821
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 204 KEAKLRDLEDSLARERDTSRRLLAEKER----------EMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEE 273
Cdd:TIGR02168 822 LRERLESLERRIAATERRLEDLEEQIEElsedieslaaEIEELEELIEELESELEALLNERASLEEALALLRSELEELSE 901
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|....
gi 383792150 274 RLRlspSPTSQRSRGRASSHSSQTQGGGSVTKKRKLESTESR--SSFSQHARTSGRVAVEEVDE 335
Cdd:TIGR02168 902 ELR---ELESKRSELRRELEELREKLAQLELRLEGLEVRIDNlqERLSEEYSLTLEEAEALENK 962
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
10-240 |
5.53e-12 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 67.48 E-value: 5.53e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 10 KKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMK 89
Cdd:COG4942 24 EAEAELEQLQQEIAELEKELAALKKEEKALLKQLAALERRIAALARRIRALEQELAALEAELAELEKEIAELRAELEAQK 103
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 90 EEldfqkniYSEELRET-KRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEktysaKLDNARQSAER 168
Cdd:COG4942 104 EE-------LAELLRALyRLGRQPPLALLLSPEDFLDAVRRLQYLKYLAPARREQAEELRADLA-----ELAALRAELEA 171
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|..
gi 383792150 169 NSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARM 240
Cdd:COG4942 172 ERAELEALLAELEEERAALEALKAERQKLLARLEKELAELAAELAELQQEAEELEALIARLEAEAAAAAERT 243
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
17-289 |
9.22e-12 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 66.71 E-value: 9.22e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELDFQK 96
Cdd:COG4942 24 EAEAELEQLQQEIAELEKELAALKKEEKALLKQLAALERRIAALARRIRALEQELAALEAELAELEKEIAELRAELEAQK 103
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 97 NIYSEELRET-KRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEktysakldnarqsaernsnlvga 175
Cdd:COG4942 104 EELAELLRALyRLGRQPPLALLLSPEDFLDAVRRLQYLKYLAPARREQAEELRADLA----------------------- 160
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 176 aheelqqsriridslsaQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKL 255
Cdd:COG4942 161 -----------------ELAALRAELEAERAELEALLAELEEERAALEALKAERQKLLARLEKELAELAAELAELQQEAE 223
|
250 260 270
....*....|....*....|....*....|....
gi 383792150 256 ALDMEIhayRKLLEGEEERLRLSPSPTSQRSRGR 289
Cdd:COG4942 224 ELEALI---ARLEAEAAAAAERTPAAGFAALKGK 254
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
9-276 |
1.74e-11 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 67.39 E-value: 1.74e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 9 TKKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLeaalgEAKKQLQDEMLRRVDAEN-RLQT 87
Cdd:TIGR02168 249 KEAEEELEELTAELQELEEKLEELRLEVSELEEEIEELQKELYALANEISRL-----EQQKQILRERLANLERQLeELEA 323
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 88 MKEELDFQKNIYSEELRETKRRHETRLVEIDNgkQREFESRLADALQELraqhEDQVEQYKKELEkTYSAKLDNARQSAE 167
Cdd:TIGR02168 324 QLEELESKLDELAEELAELEEKLEELKEELES--LEAELEELEAELEEL----ESRLEELEEQLE-TLRSKVAQLELQIA 396
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 168 RNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLaaKEAKLRDLEDSLArERDTSRRLLAEKEREMAEMRARMQQQLDEY 247
Cdd:TIGR02168 397 SLNNEIERLEARLERLEDRRERLQQEIEELLKKL--EEAELKELQAELE-ELEEELEELQEELERLEEALEELREELEEA 473
|
250 260
....*....|....*....|....*....
gi 383792150 248 QELLDIKLALDMEIHAYRKLLEGEEERLR 276
Cdd:TIGR02168 474 EQALDAAERELAQLQARLDSLERLQENLE 502
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
18-282 |
4.06e-11 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 66.09 E-value: 4.06e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 18 AQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAA---------LGEAKKQLQD--EMLRRVDAEN-RL 85
Cdd:COG4913 608 NRAKLAALEAELAELEEELAEAEERLEALEAELDALQERREALQRLaeyswdeidVASAEREIAEleAELERLDASSdDL 687
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 86 QTMKEELdfqkniysEELRETKRRHETRLVEIdNGKQREFESRLADAlQELRAQHEDQVEQYKKELEKTYSAKLDnARQS 165
Cdd:COG4913 688 AALEEQL--------EELEAELEELEEELDEL-KGEIGRLEKELEQA-EEELDELQDRLEAAEDLARLELRALLE-ERFA 756
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 166 AERNSNLVGAAHEELQQsriRIDSLSAQLSQLQKQLAAK--------EAKLRDLEDSLARERDTSRRLLAEKEREMAEMR 237
Cdd:COG4913 757 AALGDAVERELRENLEE---RIDALRARLNRAEEELERAmrafnrewPAETADLDADLESLPEYLALLDRLEEDGLPEYE 833
|
250 260 270 280 290
....*....|....*....|....*....|....*....|....*....|....*...
gi 383792150 238 ARMQQQLDE--YQELLDIKLALDMEIHAYRKLLE-----------GEEERLRLSPSPT 282
Cdd:COG4913 834 ERFKELLNEnsIEFVADLLSKLRRAIREIKERIDplndslkripfGPGRYLRLEARPR 891
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
16-276 |
6.16e-11 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 65.34 E-value: 6.16e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 16 IAAQARLKDLEALLnskeAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELdFQ 95
Cdd:COG1196 218 LKEELKELEAELLL----LKLRELEAELEELEAELEELEAELEELEAELAELEAELEELRLELEELELELEEAQAEE-YE 292
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 96 KNIYSEELRETKRRHETRLveIDNGKQREfesRLADALQELRAQHEDQVEQYKKELEKtySAKLDNARQSAERNsnLVGA 175
Cdd:COG1196 293 LLAELARLEQDIARLEERR--RELEERLE---ELEEELAELEEELEELEEELEELEEE--LEEAEEELEEAEAE--LAEA 363
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 176 AHEELQQSRIRIDSLSAQLSQLQKQLAA--KEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDI 253
Cdd:COG1196 364 EEALLEAEAELAEAEEELEELAEELLEAlrAAAELAAQLEELEEAEEALLERLERLEEELEELEEALAELEEEEEEEEEA 443
|
250 260
....*....|....*....|...
gi 383792150 254 KLALDMEIHAYRKLLEGEEERLR 276
Cdd:COG1196 444 LEEAAEEEAELEEEEEALLELLA 466
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
12-239 |
4.18e-10 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 62.63 E-value: 4.18e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 12 EGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEgelhDLRGQVAKLEAALGEA-KKQLQDEMLRRVDAENRLQTMKE 90
Cdd:COG4913 220 EPDTFEAADALVEHFDDLERAHEALEDAREQIELLE----PIRELAERYAAARERLaELEYLRAALRLWFAQRRLELLEA 295
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 91 ELdfqkniysEELRETKRRHETRLVEIDNgKQREFESRLADALQELRAQHEDQVEQYKKELEKTySAKLDNARQSAERNS 170
Cdd:COG4913 296 EL--------EELRAELARLEAELERLEA-RLDALREELDELEAQIRGNGGDRLEQLEREIERL-ERELEERERRRARLE 365
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 383792150 171 NLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRAR 239
Cdd:COG4913 366 ALLAALGLPLPASAEEFAALRAEAAALLEALEEELEALEEALAEAEAALRDLRRELRELEAEIASLERR 434
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
12-273 |
8.52e-10 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 62.01 E-value: 8.52e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 12 EGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQ--DEMLRRVDAE-NRLQTM 88
Cdd:TIGR02169 687 KRELSSLQSELRRIENRLDELSQELSDASRKIGEIEKEIEQLEQEEEKLKERLEELEEDLSslEQEIENVKSElKELEAR 766
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 89 KEELDFQKNIYSEELRETKRRHETRLVEIDNGKQREFESRLADalQELRAQHEDQVEQyKKELEKTYsakLDNARQSAER 168
Cdd:TIGR02169 767 IEELEEDLHKLEEALNDLEARLSHSRIPEIQAELSKLEEEVSR--IEARLREIEQKLN-RLTLEKEY---LEKEIQELQE 840
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 169 NSNLVGAAHEELQQsriRIDSLSAQLSQLQKQLAAKEAKLRDLEDS---LARERDTSRRLLAEKEREMAEMRARMQQQLD 245
Cdd:TIGR02169 841 QRIDLKEQIKSIEK---EIENLNGKKEELEEELEELEAALRDLESRlgdLKKERDELEAQLRELERKIEELEAQIEKKRK 917
|
250 260
....*....|....*....|....*...
gi 383792150 246 EYQELLDIKLALDMEIHAYRKLLEGEEE 273
Cdd:TIGR02169 918 RLSELKAKLEALEEELSEIEDPKGEDEE 945
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
15-336 |
2.50e-09 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 60.46 E-value: 2.50e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 15 LIAAQARLKDLEALLNSKEAALSTaLSEKRTLEGELHDLRGQVAKLEAAL-GEAKKQLQDEMLRRVDAENRLQTMKEELD 93
Cdd:TIGR02168 181 LERTRENLDRLEDILNELERQLKS-LERQAEKAERYKELKAELRELELALlVLRLEELREELEELQEELKEAEEELEELT 259
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 94 FQKNIYSEELRETKRRH---ETRLVEID------NGKQREFESRLAdALQELRAQHEDQVEQYKKELEKTYSAKLDNARQ 164
Cdd:TIGR02168 260 AELQELEEKLEELRLEVselEEEIEELQkelyalANEISRLEQQKQ-ILRERLANLERQLEELEAQLEELESKLDELAEE 338
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 165 SAERNsnlvgaahEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDslarERDTSRRLLAEKEREMAEMRARMQQQL 244
Cdd:TIGR02168 339 LAELE--------EKLEELKEELESLEAELEELEAELEELESRLEELEE----QLETLRSKVAQLELQIASLNNEIERLE 406
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 245 DEYQELLDIKLALDMEIHA-YRKLLEGEEERLRLSPSPTSQRSRGRASSHSSQTQGGGSVTKKRKLESTESRSSFSQHAR 323
Cdd:TIGR02168 407 ARLERLEDRRERLQQEIEElLKKLEEAELKELQAELEELEEELEELQEELERLEEALEELREELEEAEQALDAAERELAQ 486
|
330
....*....|...
gi 383792150 324 TSGRVAVEEVDEE 336
Cdd:TIGR02168 487 LQARLDSLERLQE 499
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
77-276 |
1.03e-08 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 58.41 E-value: 1.03e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 77 RRVDAENRLQTMKEELD-------------------------FQKniYSEELREtkRRHETRLVEID--NGKQREFESRL 129
Cdd:COG1196 173 RKEEAERKLEATEENLErledilgelerqleplerqaekaerYRE--LKEELKE--LEAELLLLKLRelEAELEELEAEL 248
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 130 ADALQELR------AQHEDQVEQYKKELE------KTYSAKLDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQL 197
Cdd:COG1196 249 EELEAELEeleaelAELEAELEELRLELEelelelEEAQAEEYELLAELARLEQDIARLEERRRELEERLEELEEELAEL 328
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 198 QKQLAAKEAKLRDLEDSLAR---ERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEER 274
Cdd:COG1196 329 EEELEELEEELEELEEELEEaeeELEEAEAELAEAEEALLEAEAELAEAEEELEELAEELLEALRAAAELAAQLEELEEA 408
|
..
gi 383792150 275 LR 276
Cdd:COG1196 409 EE 410
|
|
| CALCOCO1 |
pfam07888 |
Calcium binding and coiled-coil domain (CALCOCO1) like; Proteins found in this family are ... |
9-252 |
1.36e-07 |
|
Calcium binding and coiled-coil domain (CALCOCO1) like; Proteins found in this family are similar to the coiled-coil transcriptional coactivator protein coexpressed by Mus musculus (CoCoA/CALCOCO1). This protein binds to a highly conserved N-terminal domain of p160 coactivators, such as GRIP1, and thus enhances transcriptional activation by a number of nuclear receptors. CALCOCO1 has a central coiled-coil region with three leucine zipper motifs, which is required for its interaction with GRIP1 and may regulate the autonomous transcriptional activation activity of the C-terminal region.
Pssm-ID: 462303 [Multi-domain] Cd Length: 488 Bit Score: 54.13 E-value: 1.36e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 9 TKKEGDLIAAQARLKDLEALLNSKEAALSTALSEK----RTLEGELHDLRGQVAKLEAALGEAKK-------QLQDEMLR 77
Cdd:pfam07888 93 REKHEELEEKYKELSASSEELSEEKDALLAQRAAHeariRELEEDIKTLTQRVLERETELERMKErakkagaQRKEEEAE 172
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 78 RVDAENRLQTMKEEL-----DFQKNIYSEELRETK----RRHETRLVEIDNGKQR---EFESRLAD--ALQELRAQHEDQ 143
Cdd:pfam07888 173 RKQLQAKLQQTEEELrslskEFQELRNSLAQRDTQvlqlQDTITTLTQKLTTAHRkeaENEALLEElrSLQERLNASERK 252
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 144 VEQYKKELEKTYS------AKLDNAR-QSAERNSNLVGAA-------------HEELQQS----RIRIDSLSAQLSQLQK 199
Cdd:pfam07888 253 VEGLGEELSSMAAqrdrtqAELHQARlQAAQLTLQLADASlalregrarwaqeRETLQQSaeadKDRIEKLSAELQRLEE 332
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 200 QLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQ-------QQLDEYQELLD 252
Cdd:pfam07888 333 RLQEERMEREKLEVELGREKDCNRVQLSESRRELQELKASLRvaqkekeQLQAEKQELLE 392
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
13-273 |
1.55e-07 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 54.54 E-value: 1.55e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 13 GDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQ-------DEMLRRVDAENRL 85
Cdd:COG4913 685 DDLAALEEQLEELEAELEELEEELDELKGEIGRLEKELEQAEEELDELQDRLEAAEDLARlelrallEERFAAALGDAVE 764
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 86 QTMKEELdfQKNIysEELRETKRRHETRLVEIDNGKQREFESRLAD------ALQELRAQHEDQVEQykkELEKtYSAKL 159
Cdd:COG4913 765 RELRENL--EERI--DALRARLNRAEEELERAMRAFNREWPAETADldadleSLPEYLALLDRLEED---GLPE-YEERF 836
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 160 DNARQSAERN--SNLVGAAHEELQQSRIRIDSLSAQLSQLQ------KQLAAKEAKL-------RDLEDSLARERDTSRR 224
Cdd:COG4913 837 KELLNENSIEfvADLLSKLRRAIREIKERIDPLNDSLKRIPfgpgryLRLEARPRPDpevrefrQELRAVTSGASLFDEE 916
|
250 260 270 280 290
....*....|....*....|....*....|....*....|....*....|...
gi 383792150 225 LLAEKEREMAEMRARMQQQlDEYQELLDIKLALD----MEIHAYRKLLEGEEE 273
Cdd:COG4913 917 LSEARFAALKRLIERLRSE-EEESDRRWRARVLDvrnhLEFDAEEIDREDGEE 968
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
73-278 |
1.60e-07 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 54.25 E-value: 1.60e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 73 DEMLRRVDAENRLQTMKEELDFQKNIYSEELRE---TKRRHETRLVEID-NGKQREFESRLADALQE--LRAQHEDQVEQ 146
Cdd:COG3206 93 RPVLERVVDKLNLDEDPLGEEASREAAIERLRKnltVEPVKGSNVIEISyTSPDPELAAAVANALAEayLEQNLELRREE 172
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 147 YKKELE------KTYSAKLDNARQSAE--RNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARE 218
Cdd:COG3206 173 ARKALEfleeqlPELRKELEEAEAALEefRQKNGLVDLSEEAKLLLQQLSELESQLAEARAELAEAEARLAALRAQLGSG 252
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....*...
gi 383792150 219 RDTSRRLLAekEREMAEMRARMQQQLDEYQELL--------DIKlALDMEIHAYRKLLEGEEERLRLS 278
Cdd:COG3206 253 PDALPELLQ--SPVIQQLRAQLAELEAELAELSarytpnhpDVI-ALRAQIAALRAQLQQEAQRILAS 317
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
21-276 |
2.33e-07 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 54.03 E-value: 2.33e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 21 RLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQL--------------------QDEMLRRV- 79
Cdd:pfam01576 750 QVRELEAELEDERKQRAQAVAAKKKLELDLKELEAQIDAANKGREEAVKQLkklqaqmkdlqreleearasRDEILAQSk 829
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 80 DAENRLQTMKEE-LDFQKNIYSEE--------------------------LRETKRRHETRLVEIDngKQREFESRLADA 132
Cdd:pfam01576 830 ESEKKLKNLEAElLQLQEDLAASErarrqaqqerdeladeiasgasgksaLQDEKRRLEARIAQLE--EELEEEQSNTEL 907
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 133 LQELRAQHEDQVEQYKKEL--EKTYSAKLDNARQSAERNSNLVGAAHEELQqsriridslSAQLSQLQKQLAAKEAKLRD 210
Cdd:pfam01576 908 LNDRLRKSTLQVEQLTTELaaERSTSQKSESARQQLERQNKELKAKLQEME---------GTVKSKFKSSIAALEAKIAQ 978
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|..
gi 383792150 211 LEDSL---ARERDTSRRLLAEKEREMAEMRARMQ---QQLDEYQELLDiKLALDMEiHAYRKLLEGEEERLR 276
Cdd:pfam01576 979 LEEQLeqeSRERQAANKLVRRTEKKLKEVLLQVEderRHADQYKDQAE-KGNSRMK-QLKRQLEEAEEEASR 1048
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
18-278 |
5.03e-07 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 52.73 E-value: 5.03e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 18 AQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELdfqkn 97
Cdd:PRK02224 361 LREEAAELESELEEAREAVEDRREEIEELEEEIEELRERFGDAPVDLGNAEDFLEELREERDELREREAELEATL----- 435
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 98 iysEELRETKRRHETRLveiDNGKQREFESRLADA---------------LQELRAQHEDQVEQYKKELEKTYSAK---- 158
Cdd:PRK02224 436 ---RTARERVEEAEALL---EAGKCPECGQPVEGSphvetieedrerveeLEAELEDLEEEVEEVEERLERAEDLVeaed 509
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 159 -LDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTsrrlLAEKEREMAEMR 237
Cdd:PRK02224 510 rIERLEERREDLEELIAERRETIEEKRERAEELRERAAELEAEAEEKREAAAEAEEEAEEAREE----VAELNSKLAELK 585
|
250 260 270 280
....*....|....*....|....*....|....*....|....*.
gi 383792150 238 ARMqQQLDEYQELLDIKLALDMEIHAYRKLLEG-----EEERLRLS 278
Cdd:PRK02224 586 ERI-ESLERIRTLLAAIADAEDEIERLREKREAlaelnDERRERLA 630
|
|
| CwlO1 |
COG3883 |
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function ... |
26-304 |
8.17e-07 |
|
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function unknown];
Pssm-ID: 443091 [Multi-domain] Cd Length: 379 Bit Score: 51.37 E-value: 8.17e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 26 EALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELDFQKNIYSEELRE 105
Cdd:COG3883 15 DPQIQAKQKELSELQAELEAAQAELDALQAELEELNEEYNELQAELEALQAEIDKLQAEIAEAEAEIEERREELGERARA 94
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 106 TKR--RHETRLVEIDNGKQ-REFESRLaDALQELRAQHEDQVEQYKKELEKtysakLDNARQSAERNSNLVGAAHEELQQ 182
Cdd:COG3883 95 LYRsgGSVSYLDVLLGSESfSDFLDRL-SALSKIADADADLLEELKADKAE-----LEAKKAELEAKLAELEALKAELEA 168
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 183 SRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALDMEIH 262
Cdd:COG3883 169 AKAELEAQQAEQEALLAQLSAEEAAAEAQLAELEAELAAAEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAASAAG 248
|
250 260 270 280
....*....|....*....|....*....|....*....|..
gi 383792150 263 AYRKLLEGEEERLRLSPSPTSQRSRGRASSHSSQTQGGGSVT 304
Cdd:COG3883 249 AGAAGAAGAAAGSAGAAGAAAGAAGAGAAAASAAGGGAGGAG 290
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
19-243 |
9.26e-07 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 51.94 E-value: 9.26e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 19 QARLKDLEALLNSKEAAL----------------STALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQ--DEMLRRVD 80
Cdd:COG3206 181 EEQLPELRKELEEAEAALeefrqknglvdlseeaKLLLQQLSELESQLAEARAELAEAEARLAALRAQLGsgPDALPELL 260
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 81 AENRLQTMKEELdfqkniyseelretkrrhetrlveidngkqREFESRLADALQELRAQHEDqVEQYKKELEKTysakld 160
Cdd:COG3206 261 QSPVIQQLRAQL------------------------------AELEAELAELSARYTPNHPD-VIALRAQIAAL------ 303
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 161 nARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQ---LAAKEAKLRDLEdslaRERDTSRRLLAEKEREMAEMR 237
Cdd:COG3206 304 -RAQLQQEAQRILASLEAELEALQAREASLQAQLAQLEARlaeLPELEAELRRLE----REVEVARELYESLLQRLEEAR 378
|
....*.
gi 383792150 238 ARMQQQ 243
Cdd:COG3206 379 LAEALT 384
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
19-250 |
1.48e-06 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 51.19 E-value: 1.48e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 19 QARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQ-----LQDEML--RRVDAENRLQTMKEE 91
Cdd:PRK02224 250 REELETLEAEIEDLRETIAETEREREELAEEVRDLRERLEELEEERDDLLAEaglddADAEAVeaRREELEDRDEELRDR 329
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 92 LDFQK------NIYSEELRETKRRHETRLVEIDN----------------GKQREFESRLADALQELRAQHED------Q 143
Cdd:PRK02224 330 LEECRvaaqahNEEAESLREDADDLEERAEELREeaaeleseleeareavEDRREEIEELEEEIEELRERFGDapvdlgN 409
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 144 VEQYKKEL----------EKTYSAKLDNARQSAERNSNLV-------------GAAH-EELQQSRIRIDSLSAQLSQLQK 199
Cdd:PRK02224 410 AEDFLEELreerdelrerEAELEATLRTARERVEEAEALLeagkcpecgqpveGSPHvETIEEDRERVEELEAELEDLEE 489
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 200 QLAAKEAKLRDLED---------SLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQEL 250
Cdd:PRK02224 490 EVEEVEERLERAEDlveaedrieRLEERREDLEELIAERRETIEEKRERAEELRERAAEL 549
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
118-276 |
2.08e-06 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 50.68 E-value: 2.08e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 118 DNGKQREFESRLADALQELRAQHEDQVEQYKKELEkTYSAKLDNARQSAERNSNL--VGAAHEELQQSRIRIDSL---SA 192
Cdd:COG4913 607 DNRAKLAALEAELAELEEELAEAEERLEALEAELD-ALQERREALQRLAEYSWDEidVASAEREIAELEAELERLdasSD 685
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 193 QLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEReMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEE 272
Cdd:COG4913 686 DLAALEEQLEELEAELEELEEELDELKGEIGRLEKELEQ-AEEELDELQDRLEAAEDLARLELRALLEERFAAALGDAVE 764
|
....
gi 383792150 273 ERLR 276
Cdd:COG4913 765 RELR 768
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
15-177 |
2.57e-06 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 48.77 E-value: 2.57e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 15 LIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQ-----LQDEM----LRRVDAENRL 85
Cdd:COG1579 33 LAELEDELAALEARLEAAKTELEDLEKEIKRLELEIEEVEARIKKYEEQLGNVRNNkeyeaLQKEIeslkRRISDLEDEI 112
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 86 QTMKEELDFQKniysEELRETKRRHETRLVEIDNgKQREFESRLADaLQELRAQHEDQVEQYKKELEKTYSAKLDNARqs 165
Cdd:COG1579 113 LELMERIEELE----EELAELEAELAELEAELEE-KKAELDEELAE-LEAELEELEAEREELAAKIPPELLALYERIR-- 184
|
170
....*....|..
gi 383792150 166 aeRNSNLVGAAH 177
Cdd:COG1579 185 --KRKNGLAVVP 194
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
9-249 |
2.62e-06 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 50.42 E-value: 2.62e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 9 TKKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQD-EMLRR--------- 78
Cdd:PRK02224 380 EDRREEIEELEEEIEELRERFGDAPVDLGNAEDFLEELREERDELREREAELEATLRTARERVEEaEALLEagkcpecgq 459
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 79 -VDAENRLQTMkEELDFQKNIYSEELRETKRRHETRLVEIDNGKQ-REFESRL------ADALQELRAQHEDQVEQYKKE 150
Cdd:PRK02224 460 pVEGSPHVETI-EEDRERVEELEAELEDLEEEVEEVEERLERAEDlVEAEDRIerleerREDLEELIAERRETIEEKRER 538
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 151 LE--KTYSAKLDNARQSAERNSNlvgAAHEELQQSRIRIDSLSAQLSQLQKQLAAkeakLRDLEDSLARERDTSRRL--L 226
Cdd:PRK02224 539 AEelRERAAELEAEAEEKREAAA---EAEEEAEEAREEVAELNSKLAELKERIES----LERIRTLLAAIADAEDEIerL 611
|
250 260
....*....|....*....|...
gi 383792150 227 AEKEREMAEMRARMQQQLDEYQE 249
Cdd:PRK02224 612 REKREALAELNDERRERLAEKRE 634
|
|
| MukB |
COG3096 |
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell ... |
27-236 |
2.96e-06 |
|
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 442330 [Multi-domain] Cd Length: 1470 Bit Score: 50.34 E-value: 2.96e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 27 ALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQdeMLRRV----------DAENRLQTMKEELDFqk 96
Cdd:COG3096 829 AFAPDPEAELAALRQRRSELERELAQHRAQEQQLRQQLDQLKEQLQ--LLNKLlpqanlladeTLADRLEELREELDA-- 904
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 97 niySEELRETKRRHETRLVEIDngkqrefesRLADALQELRAQHEDQVEQYK--KELEKTYSAKLDNARQSAERNSNLVG 174
Cdd:COG3096 905 ---AQEAQAFIQQHGKALAQLE---------PLVAVLQSDPEQFEQLQADYLqaKEQQRRLKQQIFALSEVVQRRPHFSY 972
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 383792150 175 A-AHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLED----------SLARERDTSRRLLAEKEREMAEM 236
Cdd:COG3096 973 EdAVGLLGENSDLNEKLRARLEQAEEARREAREQLRQAQAqysqynqvlaSLKSSRDAKQQTLQELEQELEEL 1045
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
49-250 |
3.01e-06 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 48.77 E-value: 3.01e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 49 ELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELdfqkniysEELRETKRRHETRLVEIDNgKQREFESR 128
Cdd:COG1579 11 DLQELDSELDRLEHRLKELPAELAELEDELAALEARLEAAKTEL--------EDLEKEIKRLELEIEEVEA-RIKKYEEQ 81
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 129 LAdalqelRAQHEDQVEQYKKELEKtysakLDNARQSAErnsnlvgaahEELQQSRIRIDSLSAQLSQLQKQLAAKEAKL 208
Cdd:COG1579 82 LG------NVRNNKEYEALQKEIES-----LKRRISDLE----------DEILELMERIEELEEELAELEAELAELEAEL 140
|
170 180 190 200
....*....|....*....|....*....|....*....|...
gi 383792150 209 RDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQL-DEYQEL 250
Cdd:COG1579 141 EEKKAELDEELAELEAELEELEAEREELAAKIPPELlALYERI 183
|
|
| PRK11281 |
PRK11281 |
mechanosensitive channel MscK; |
19-254 |
3.56e-06 |
|
mechanosensitive channel MscK;
Pssm-ID: 236892 [Multi-domain] Cd Length: 1113 Bit Score: 50.30 E-value: 3.56e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 19 QARLKDL--EALLNSKEAALSTALSEkrTLegelhDLRGQVAKLEAALGEAKKQLQDemlrrvdAENRLQTMKEELDFQK 96
Cdd:PRK11281 42 QAQLDALnkQKLLEAEDKLVQQDLEQ--TL-----ALLDKIDRQKEETEQLKQQLAQ-------APAKLRQAQAELEALK 107
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 97 NIYSEELRETkrrhetrlveIDNGKQREFESRLADALQELraqhedqvEQYKKELEkTYSAKLDNARQSAERNSNLVGAA 176
Cdd:PRK11281 108 DDNDEETRET----------LSTLSLRQLESRLAQTLDQL--------QNAQNDLA-EYNSQLVSLQTQPERAQAALYAN 168
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 177 HEELQQSRIRIDSLSA--------QLSQLQKQLAAKEAKLrDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQ 248
Cdd:PRK11281 169 SQRLQQIRNLLKGGKVggkalrpsQRVLLQAEQALLNAQN-DLQRKSLEGNTQLQDLLQKQRDYLTARIQRLEHQLQLLQ 247
|
....*.
gi 383792150 249 ELLDIK 254
Cdd:PRK11281 248 EAINSK 253
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
8-207 |
3.86e-06 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 49.77 E-value: 3.86e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 8 NTKKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALgeakkQLQDEMLRRVDAENRLQT 87
Cdd:COG4717 69 NLKELKELEEELKEAEEKEEEYAELQEELEELEEELEELEAELEELREELEKLEKLL-----QLLPLYQELEALEAELAE 143
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 88 MKEELdfqkniysEELRETKRRHETRLVEIDNGKQ--REFESRLADALQELRAQHEDQVEQYKKELEktysaKLDNARQS 165
Cdd:COG4717 144 LPERL--------EELEERLEELRELEEELEELEAelAELQEELEELLEQLSLATEEELQDLAEELE-----ELQQRLAE 210
|
170 180 190 200
....*....|....*....|....*....|....*....|..
gi 383792150 166 AErnsnlvgaahEELQQSRIRIDSLSAQLSQLQKQLAAKEAK 207
Cdd:COG4717 211 LE----------EELEEAQEELEELEEELEQLENELEAAALE 242
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
20-269 |
5.11e-06 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 49.68 E-value: 5.11e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 20 ARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAA--LGEAKKQLQDEMLRRVDAENR-LQTMKEELDFQK 96
Cdd:PRK03918 459 AELKRIEKELKEIEEKERKLRKELRELEKVLKKESELIKLKELAeqLKELEEKLKKYNLEELEKKAEeYEKLKEKLIKLK 538
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 97 NIYSEELRETKRRHE--TRLVEIDNgKQREFESRLADALQELRA---QHEDQVEQYKKELEKTYSA--KLDNARQSAERN 169
Cdd:PRK03918 539 GEIKSLKKELEKLEElkKKLAELEK-KLDELEEELAELLKELEElgfESVEELEERLKELEPFYNEylELKDAEKELERE 617
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 170 SNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTsrrllaEKEREMAEMRARMQQ---QLDE 246
Cdd:PRK03918 618 EKELKKLEEELDKAFEELAETEKRLEELRKELEELEKKYSEEEYEELREEYL------ELSRELAGLRAELEElekRREE 691
|
250 260
....*....|....*....|...
gi 383792150 247 YQELLDIKLALDMEIHAYRKLLE 269
Cdd:PRK03918 692 IKKTLEKLKEELEEREKAKKELE 714
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
77-336 |
6.98e-06 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 49.28 E-value: 6.98e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 77 RRVDAENRLQTMKEELDFQKNI-----------------------YSEELRET-------------KRRHETRLVEIDNG 120
Cdd:TIGR02168 173 RRKETERKLERTRENLDRLEDIlnelerqlkslerqaekaerykeLKAELRELelallvlrleelrEELEELQEELKEAE 252
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 121 KQREFESRLADALQELRAQHEDQVEQYKKELEKtYSAKLDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQ 200
Cdd:TIGR02168 253 EELEELTAELQELEEKLEELRLEVSELEEEIEE-LQKELYALANEISRLEQQKQILRERLANLERQLEELEAQLEELESK 331
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 201 LAAKEAKLRDLE----------DSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEG 270
Cdd:TIGR02168 332 LDELAEELAELEekleelkeelESLEAELEELEAELEELESRLEELEEQLETLRSKVAQLELQIASLNNEIERLEARLER 411
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 383792150 271 EEERLRLSPSPTSQRSRgRASSHSSQTQGGGSVTKKRKLESTESRSSFSQHARTSGRVAVEEVDEE 336
Cdd:TIGR02168 412 LEDRRERLQQEIEELLK-KLEEAELKELQAELEELEEELEELQEELERLEEALEELREELEEAEQA 476
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
41-275 |
8.70e-06 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 48.91 E-value: 8.70e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 41 SEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAEnRLQTMKEELdfQKNIYSEELREtKRRHETRLVEIDNg 120
Cdd:TIGR02169 170 RKKEKALEELEEVEENIERLDLIIDEKRQQLERLRREREKAE-RYQALLKEK--REYEGYELLKE-KEALERQKEAIER- 244
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 121 kqrefesRLADALQELraqheDQVEQYKKELEKTYSAKLDNARQSAERNSNLVGaahEELQQSRIRIDSLSAQLSQL--- 197
Cdd:TIGR02169 245 -------QLASLEEEL-----EKLTEEISELEKRLEEIEQLLEELNKKIKDLGE---EEQLRVKEKIGELEAEIASLers 309
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 198 -------QKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEG 270
Cdd:TIGR02169 310 iaekereLEDAEERLAKLEAEIDKLLAEIEELEREIEEERKRRDKLTEEYAELKEELEDLRAELEEVDKEFAETRDELKD 389
|
....*
gi 383792150 271 EEERL 275
Cdd:TIGR02169 390 YREKL 394
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
10-277 |
1.24e-05 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 48.14 E-value: 1.24e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 10 KKEGDLIAAQARLKDLEAL---LNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQ 86
Cdd:PRK03918 218 ELREELEKLEKEVKELEELkeeIEELEKELESLEGSKRKLEEKIRELEERIEELKKEIEELEEKVKELKELKEKAEEYIK 297
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 87 TMKEELDFQKNIYSEELRETKRRH-----ETRLVEIDNGKQR-----EFESRLADALQELRAQHE--DQVEQYKKELEKT 154
Cdd:PRK03918 298 LSEFYEEYLDELREIEKRLSRLEEeingiEERIKELEEKEERleelkKKLKELEKRLEELEERHElyEEAKAKKEELERL 377
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 155 YSAKLDNARQSAERNSNLVGAAHEELQQsriRIDSLSAQLSQLQKQLAAKEAKLRDLEdSLARERDTSRRLLAEKERE-- 232
Cdd:PRK03918 378 KKRLTGLTPEKLEKELEELEKAKEEIEE---EISKITARIGELKKEIKELKKAIEELK-KAKGKCPVCGRELTEEHRKel 453
|
250 260 270 280
....*....|....*....|....*....|....*....|....*
gi 383792150 233 MAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEERLRL 277
Cdd:PRK03918 454 LEEYTAELKRIEKELKEIEEKERKLRKELRELEKVLKKESELIKL 498
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
20-276 |
1.29e-05 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 48.25 E-value: 1.29e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 20 ARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEE---LDFQK 96
Cdd:pfam01576 68 ARKQELEEILHELESRLEEEEERSQQLQNEKKKMQQHIQDLEEQLDEEEAARQKLQLEKVTTEAKIKKLEEDillLEDQN 147
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 97 NIYSEElretKRRHETRLVEIDNGKQREFESrlADALQELRAQHEDQVEQYKKELEKTysaklDNARQSAERNSNLVGAA 176
Cdd:pfam01576 148 SKLSKE----RKLLEERISEFTSNLAEEEEK--AKSLSKLKNKHEAMISDLEERLKKE-----EKGRQELEKAKRKLEGE 216
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 177 HEELQQsriRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERdTSRRLLAEKEREmaemrarMQQQLDEYQELLDIKLA 256
Cdd:pfam01576 217 STDLQE---QIAELQAQIAELRAQLAKKEEELQAALARLEEET-AQKNNALKKIRE-------LEAQISELQEDLESERA 285
|
250 260
....*....|....*....|
gi 383792150 257 LDMEIHAYRKLLEGEEERLR 276
Cdd:pfam01576 286 ARNKAEKQRRDLGEELEALK 305
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
43-276 |
1.46e-05 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 48.09 E-value: 1.46e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 43 KRTLEGELHDLRGQVAKLEAALGEAKKQLQDemlrrvdAENRLQTMKEeldfQKNIYSeeLRETKRRHETRLVEIdngkq 122
Cdd:COG3206 163 EQNLELRREEARKALEFLEEQLPELRKELEE-------AEAALEEFRQ----KNGLVD--LSEEAKLLLQQLSEL----- 224
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 123 refESRLADAlQELRAQHEDQVEQYKKELEKTYSAkldnarQSAERNSNLVGAAHEELQQSRIRIDSLSAQLS------- 195
Cdd:COG3206 225 ---ESQLAEA-RAELAEAEARLAALRAQLGSGPDA------LPELLQSPVIQQLRAQLAELEAELAELSARYTpnhpdvi 294
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 196 QLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEERL 275
Cdd:COG3206 295 ALRAQIAALRAQLQQEAQRILASLEAELEALQAREASLQAQLAQLEARLAELPELEAELRRLEREVEVARELYESLLQRL 374
|
.
gi 383792150 276 R 276
Cdd:COG3206 375 E 375
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
14-233 |
3.14e-05 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 47.22 E-value: 3.14e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 14 DLIAAQARLKDLEALLNSkeAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDemlrrvdAENRLQTMKEELD 93
Cdd:COG4913 263 RYAAARERLAELEYLRAA--LRLWFAQRRLELLEAELEELRAELARLEAELERLEARLDA-------LREELDELEAQIR 333
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 94 FQKNIYSEELRETKRRHETRLVEIdngkQREFEsRLADALQELRAQHEDQVEQYKkelektysAKLDNARQSAERNSNLV 173
Cdd:COG4913 334 GNGGDRLEQLEREIERLERELEER----ERRRA-RLEALLAALGLPLPASAEEFA--------ALRAEAAALLEALEEEL 400
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 174 GAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREM 233
Cdd:COG4913 401 EALEEALAEAEAALRDLRRELRELEAEIASLERRKSNIPARLLALRDALAEALGLDEAEL 460
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
16-213 |
4.70e-05 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 46.30 E-value: 4.70e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 16 IAAQARLKDLEALLNSKEAALStalsEKRTLEGELHDLRGQVAKLEAALGEAKKQLqdEMLRRVDAENRLQTMKEELDFQ 95
Cdd:COG4717 67 ELNLKELKELEEELKEAEEKEE----EYAELQEELEELEEELEELEAELEELREEL--EKLEKLLQLLPLYQELEALEAE 140
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 96 KNIYSEELRETKRRHEtrlveidngkqrefesRLADALQELRAQhEDQVEQYKKELEKTYSAKLDNARQSAERNSNLVGA 175
Cdd:COG4717 141 LAELPERLEELEERLE----------------ELRELEEELEEL-EAELAELQEELEELLEQLSLATEEELQDLAEELEE 203
|
170 180 190
....*....|....*....|....*....|....*...
gi 383792150 176 AHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLED 213
Cdd:COG4717 204 LQQRLAELEEELEEAQEELEELEEELEQLENELEAAAL 241
|
|
| COG1340 |
COG1340 |
Uncharacterized coiled-coil protein, contains DUF342 domain [Function unknown]; |
41-239 |
5.27e-05 |
|
Uncharacterized coiled-coil protein, contains DUF342 domain [Function unknown];
Pssm-ID: 440951 [Multi-domain] Cd Length: 297 Bit Score: 45.29 E-value: 5.27e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 41 SEKRTLEGELHDLRGQVAKLEAALGEAKKQLQD--EMLRRVDA-ENRLQTMKEELDFQKNIYsEELRETKRRHETRLVEI 117
Cdd:COG1340 78 EERDELNEKLNELREELDELRKELAELNKAGGSidKLRKEIERlEWRQQTEVLSPEEEKELV-EKIKELEKELEKAKKAL 156
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 118 DNGKQREFESRLADALQELRAQHEDQVEQYKKELEKtYSAKLDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQL 197
Cdd:COG1340 157 EKNEKLKELRAELKELRKEAEEIHKKIKELAEEAQE-LHEEMIELYKEADELRKEADELHKEIVEAQEKADELHEEIIEL 235
|
170 180 190 200
....*....|....*....|....*....|....*....|..
gi 383792150 198 QKqlaakeaKLRDLEDSLARERDTSRRLLAEKEREMAEMRAR 239
Cdd:COG1340 236 QK-------ELRELRKELKKLRKKQRALKREKEKEELEEKAE 270
|
|
| Crescentin |
pfam19220 |
Crescentin protein; This entry represents a bacterial equivalent to Intermediate Filament ... |
20-245 |
5.42e-05 |
|
Crescentin protein; This entry represents a bacterial equivalent to Intermediate Filament proteins, named crescentin, whose cytoskeletal function is required for the vibrioid and helical shapes of Caulobacter crescentus. Without crescentin, the cells adopt a straight-rod morphology. Crescentin has characteriztic features of IF proteins including the ability to assemble into filaments in vitro without energy or cofactor requirements. In vivo, crescentin forms a helical structure that colocalizes with the inner cell curvatures beneath the cytoplasmic membrane.
Pssm-ID: 437057 [Multi-domain] Cd Length: 401 Bit Score: 45.83 E-value: 5.42e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 20 ARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDE---MLRRVDA-ENRLQTMKE---EL 92
Cdd:pfam19220 188 AELAELTRRLAELETQLDATRARLRALEGQLAAEQAERERAEAQLEEAVEAHRAErasLRMKLEAlTARAAATEQllaEA 267
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 93 DFQKNIYSEELRETKRRHETRLVEIDNgkqreFESRLAdalqELRAQHEDQVEQYKKelektysakLDNARQSAERNSNL 172
Cdd:pfam19220 268 RNQLRDRDEAIRAAERRLKEASIERDT-----LERRLA----GLEADLERRTQQFQE---------MQRARAELEERAEM 329
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*..
gi 383792150 173 VG---AAHE-ELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDledslarerdTSRRLLAEKEREMAEmRARMQQQLD 245
Cdd:pfam19220 330 LTkalAAKDaALERAEERIASLSDRIAELTKRFEVERAALEQ----------ANRRLKEELQRERAE-RALAQGALE 395
|
|
| ClpA |
COG0542 |
ATP-dependent Clp protease, ATP-binding subunit ClpA [Posttranslational modification, protein ... |
184-276 |
6.65e-05 |
|
ATP-dependent Clp protease, ATP-binding subunit ClpA [Posttranslational modification, protein turnover, chaperones];
Pssm-ID: 440308 [Multi-domain] Cd Length: 836 Bit Score: 45.84 E-value: 6.65e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 184 RIRIDSLSAQLSQLQKQLAA----KEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALD- 258
Cdd:COG0542 403 RMEIDSKPEELDELERRLEQleieKEALKKEQDEASFERLAELRDELAELEEELEALKARWEAEKELIEEIQELKEELEq 482
|
90 100
....*....|....*....|
gi 383792150 259 --MEIHAYRKLLEGEEERLR 276
Cdd:COG0542 483 ryGKIPELEKELAELEEELA 502
|
|
| Mitofilin |
pfam09731 |
Mitochondrial inner membrane protein; Mitofilin controls mitochondrial cristae morphology. ... |
17-249 |
1.12e-04 |
|
Mitochondrial inner membrane protein; Mitofilin controls mitochondrial cristae morphology. Mitofilin is enriched in the narrow space between the inner boundary and the outer membranes, where it forms a homotypic interaction and assembles into a large multimeric protein complex. The first 78 amino acids contain a typical amino-terminal-cleavable mitochondrial presequence rich in positive-charged and hydroxylated residues and a membrane anchor domain. In addition, it has three centrally located coiled coil domains.
Pssm-ID: 430783 [Multi-domain] Cd Length: 618 Bit Score: 45.13 E-value: 1.12e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARLKDLEALLNSKEAALSTALSEKRTLEGElhdlrgQVAKLEAALGEAKKQLQDEM-LRRVDAENRLQTMKEELDFQ 95
Cdd:pfam09731 129 ALEEVLKEAISKAESATAVAKEAKDDAIQAVKA------HTDSLKEASDTAEISREKATdSALQKAEALAEKLKEVINLA 202
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 96 KNIYSEELRETK-RRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVE----QYKKELEKTY-----SAKLDNARQS 165
Cdd:pfam09731 203 KQSEEEAAPPLLdAAPETPPKLPEHLDNVEEKVEKAQSLAKLVDQYKELVAseriVFQQELVSIFpdiipVLKEDNLLSN 282
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 166 AERNSnLVGAAHEELQQSRIRIDSLSAQLSQ-LQKQLAAKEAKLRDLEDSLARE-----RDTSRRLLAEKEREMAEMRAR 239
Cdd:pfam09731 283 DDLNS-LIAHAHREIDQLSKKLAELKKREEKhIERALEKQKEELDKLAEELSARleevrAADEAQLRLEFEREREEIRES 361
|
250
....*....|
gi 383792150 240 MQQQLDEYQE 249
Cdd:pfam09731 362 YEEKLRTELE 371
|
|
| mukB |
PRK04863 |
chromosome partition protein MukB; |
33-292 |
1.32e-04 |
|
chromosome partition protein MukB;
Pssm-ID: 235316 [Multi-domain] Cd Length: 1486 Bit Score: 44.95 E-value: 1.32e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 33 EAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQ--DEMLRRVD--AENRLQTMKEELDFQkniySEELRETKR 108
Cdd:PRK04863 836 EAELRQLNRRRVELERALADHESQEQQQRSQLEQAKEGLSalNRLLPRLNllADETLADRVEEIREQ----LDEAEEAKR 911
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 109 ---RHETRLVEIDngkqrefesRLADALQELRAQHeDQVEQYKKELEKTYSAKLDNARQSAERNSNLVGAAHEELQQSRI 185
Cdd:PRK04863 912 fvqQHGNALAQLE---------PIVSVLQSDPEQF-EQLKQDYQQAQQTQRDAKQQAFALTEVVQRRAHFSYEDAAEMLA 981
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 186 RIDSLSAQLSQLQKQLAAKEAKLRD-LEDSLARERDTSRRLLAEKERemaemRARMQQQLDEY-QELLDIKLALDmeiha 263
Cdd:PRK04863 982 KNSDLNEKLRQRLEQAEQERTRAREqLRQAQAQLAQYNQVLASLKSS-----YDAKRQMLQELkQELQDLGVPAD----- 1051
|
250 260 270
....*....|....*....|....*....|..
gi 383792150 264 yrkllEGEEERLRLSPSPTSQR---SRGRASS 292
Cdd:PRK04863 1052 -----SGAEERARARRDELHARlsaNRSRRNQ 1078
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
12-166 |
1.34e-04 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 45.01 E-value: 1.34e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 12 EGDLIAAQARLKDLEALLNSKEAALSTALSEKRTL--EGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMK 89
Cdd:COG3206 225 ESQLAEARAELAEAEARLAALRAQLGSGPDALPELlqSPVIQQLRAQLAELEAELAELSARYTPNHPDVIALRAQIAALR 304
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 90 EELDFQKNIYSEELRETKRRHETRLVEIdNGKQREFESRLADA------LQELRAQHEDQVEQYKKELEKTYSAKLDNAR 163
Cdd:COG3206 305 AQLQQEAQRILASLEAELEALQAREASL-QAQLAQLEARLAELpeleaeLRRLEREVEVARELYESLLQRLEEARLAEAL 383
|
...
gi 383792150 164 QSA 166
Cdd:COG3206 384 TVG 386
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
61-250 |
1.48e-04 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 44.76 E-value: 1.48e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 61 EAALGEAKKQLQDEMLRR------VDAENRLQTMKEELDFQKNIYSEELRETKRRHETRLVEIDNGKQREFESRLADALQ 134
Cdd:COG4717 306 ELQALPALEELEEEELEEllaalgLPPDLSPEELLELLDRIEELQELLREAEELEEELQLEELEQEIAALLAEAGVEDEE 385
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 135 ELRAQHEdQVEQYKKELEK--TYSAKLDNARQSAERNSNLVGAA--HEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRD 210
Cdd:COG4717 386 ELRAALE-QAEEYQELKEEleELEEQLEELLGELEELLEALDEEelEEELEELEEELEELEEELEELREELAELEAELEQ 464
|
170 180 190 200
....*....|....*....|....*....|....*....|
gi 383792150 211 LEDSlarerdtsrRLLAEKEREMAEMRARMQQQLDEYQEL 250
Cdd:COG4717 465 LEED---------GELAELLQELEELKAELRELAEEWAAL 495
|
|
| KpsE |
COG3524 |
Capsule polysaccharide export protein KpsE/RkpR [Cell wall/membrane/envelope biogenesis]; |
141-259 |
2.03e-04 |
|
Capsule polysaccharide export protein KpsE/RkpR [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442746 [Multi-domain] Cd Length: 370 Bit Score: 43.69 E-value: 2.03e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 141 EDQVEQYKKELEKTYsAKLDNARQsaernsnlvgaAHEELQQSRIRID------SLSAQLSQLQKQLAAKEAKLRDLEDS 214
Cdd:COG3524 176 EDAVRFAEEEVERAE-ERLRDARE-----------ALLAFRNRNGILDpeataeALLQLIATLEGQLAELEAELAALRSY 243
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*...
gi 383792150 215 L---ARERDTSRRLLAEKEREMAEMRARM---------QQQLDEYQEL-LDIKLALDM 259
Cdd:COG3524 244 LspnSPQVRQLRRRIAALEKQIAAERARLtgasggdslASLLAEYERLeLEREFAEKA 301
|
|
| DUF5401 |
pfam17380 |
Family of unknown function (DUF5401); This is a family of unknown function found in ... |
54-341 |
2.06e-04 |
|
Family of unknown function (DUF5401); This is a family of unknown function found in Chromadorea.
Pssm-ID: 375164 [Multi-domain] Cd Length: 722 Bit Score: 44.34 E-value: 2.06e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 54 RGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKE----ELDFQKNIYSEELR---ETKR-----RHETRLVEIDNGK 121
Cdd:pfam17380 287 RQQQEKFEKMEQERLRQEKEEKAREVERRRKLEEAEKarqaEMDRQAAIYAEQERmamEREReleriRQEERKRELERIR 366
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 122 QREFESRLAD--ALQELRAQHEDQVEQYKKELEKTYSAKLdnarQSAERnsnlvgaaHEELQQSRIRIDSLSAQlsqlqk 199
Cdd:pfam17380 367 QEEIAMEISRmrELERLQMERQQKNERVRQELEAARKVKI----LEEER--------QRKIQQQKVEMEQIRAE------ 428
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 200 QLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRA----RMQQQLDEYQELLDIKLA-------LDMEIHAYRKLL 268
Cdd:pfam17380 429 QEEARQREVRRLEEERAREMERVRLEEQERQQQVERLRQqeeeRKRKKLELEKEKRDRKRAeeqrrkiLEKELEERKQAM 508
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 383792150 269 EGEEERLRLSPSPTSQRSRGRASSHSSQTqgggSVTKKRKLESTESRSSFSQHAR--TSGRVAVEEVDEEGKFVR 341
Cdd:pfam17380 509 IEEERKRKLLEKEMEERQKAIYEEERRRE----AEEERRKQQEMEERRRIQEQMRkaTEERSRLEAMEREREMMR 579
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
45-276 |
2.20e-04 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 44.29 E-value: 2.20e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 45 TLEGELHDLRGQV--AKLEAALGEAKKQLQDEMLRRVDAEnrlqtmKEELDFQKNIYSEELRETKRR-HE-TRLVEIDNG 120
Cdd:TIGR02169 643 TLEGELFEKSGAMtgGSRAPRGGILFSRSEPAELQRLRER------LEGLKRELSSLQSELRRIENRlDElSQELSDASR 716
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 121 KQREFESRLADALQELRAQHEdQVEQYKKELEKTySAKLDNARQSaernsnlvgaaheelqqsrirIDSLSAQLSQLQKQ 200
Cdd:TIGR02169 717 KIGEIEKEIEQLEQEEEKLKE-RLEELEEDLSSL-EQEIENVKSE---------------------LKELEARIEELEED 773
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 201 LAAKEAKLRDLEDSLARER-DTSRRLLAEKEREMAEMRARMQ-------------QQL-DEYQELLDIKLALDMEIHAYR 265
Cdd:TIGR02169 774 LHKLEEALNDLEARLSHSRiPEIQAELSKLEEEVSRIEARLReieqklnrltlekEYLeKEIQELQEQRIDLKEQIKSIE 853
|
250
....*....|.
gi 383792150 266 KLLEGEEERLR 276
Cdd:TIGR02169 854 KEIENLNGKKE 864
|
|
| Cast |
pfam10174 |
RIM-binding protein of the cytomatrix active zone; This is a family of proteins that form part ... |
17-237 |
2.72e-04 |
|
RIM-binding protein of the cytomatrix active zone; This is a family of proteins that form part of the CAZ (cytomatrix at the active zone) complex which is involved in determining the site of synaptic vesicle fusion. The C-terminus is a PDZ-binding motif that binds directly to RIM (a small G protein Rab-3A effector). The family also contains four coiled-coil domains.
Pssm-ID: 431111 [Multi-domain] Cd Length: 766 Bit Score: 44.04 E-value: 2.72e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRG--------------QVAKLEAALGEAKKQLqDEMLRRV--- 79
Cdd:pfam10174 349 ALRLRLEEKESFLNKKTKQLQDLTEEKSTLAGEIRDLKDmldvkerkinvlqkKIENLQEQLRDKDKQL-AGLKERVksl 427
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 80 --DAENR---LQTMKEELDFQKNIYsEELRETKRRHE-TRLVEIDNGKQrefesRLADALQELRAQHEDQVEQYKKELEK 153
Cdd:pfam10174 428 qtDSSNTdtaLTTLEEALSEKERII-ERLKEQREREDrERLEELESLKK-----ENKDLKEKVSALQPELTEKESSLIDL 501
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 154 TYSAKldNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEA------KLRDLEDSLARERDTSRRLLA 227
Cdd:pfam10174 502 KEHAS--SLASSGLKKDSKLKSLEIAVEQKKEECSKLENQLKKAHNAEEAVRTnpeindRIRLLEQEVARYKEESGKAQA 579
|
250
....*....|
gi 383792150 228 EKEREMAEMR 237
Cdd:pfam10174 580 EVERLLGILR 589
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
133-278 |
3.23e-04 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 43.60 E-value: 3.23e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 133 LQELRAQHEDQVEQYKKELEKTYSAK---LDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLR 209
Cdd:COG4717 40 LAFIRAMLLERLEKEADELFKPQGRKpelNLKELKELEEELKEAEEKEEEYAELQEELEELEEELEELEAELEELREELE 119
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|..
gi 383792150 210 DLEDSLARERDTSRRllAEKEREMAEMRARMQ---QQLDEYQELLDIKLALDMEIHAYRKLLEGEEERLRLS 278
Cdd:COG4717 120 KLEKLLQLLPLYQEL--EALEAELAELPERLEeleERLEELRELEEELEELEAELAELQEELEELLEQLSLA 189
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
24-275 |
3.41e-04 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 43.51 E-value: 3.41e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 24 DLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEML--RRVDAENRLQTMKEELDFQKNIYSE 101
Cdd:PRK03918 388 KLEKELEELEKAKEEIEEEISKITARIGELKKEIKELKKAIEELKKAKGKCPVcgRELTEEHRKELLEEYTAELKRIEKE 467
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 102 --ELRETKRRHETRLVEIDNGKQREfesrladalQELRAQHE--DQVEQYKKELEKTYSAKLdnarqsaERNSNLVGAAH 177
Cdd:PRK03918 468 lkEIEEKERKLRKELRELEKVLKKE---------SELIKLKElaEQLKELEEKLKKYNLEEL-------EKKAEEYEKLK 531
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 178 EELQQSRIRIDSLSAQLSQLQ---KQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIK 254
Cdd:PRK03918 532 EKLIKLKGEIKSLKKELEKLEelkKKLAELEKKLDELEEELAELLKELEELGFESVEELEERLKELEPFYNEYLELKDAE 611
|
250 260
....*....|....*....|.
gi 383792150 255 laldMEIHAYRKLLEGEEERL 275
Cdd:PRK03918 612 ----KELEREEKELKKLEEEL 628
|
|
| CCDC158 |
pfam15921 |
Coiled-coil domain-containing protein 158; CCDC158 is a family of proteins found in eukaryotes. ... |
4-336 |
4.79e-04 |
|
Coiled-coil domain-containing protein 158; CCDC158 is a family of proteins found in eukaryotes. The function is not known.
Pssm-ID: 464943 [Multi-domain] Cd Length: 1112 Bit Score: 43.18 E-value: 4.79e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 4 SEGCNTKKEGDLIAAQAR---------LKDLEALLNSKEAALSTAlseKRTLEGELHDLRGQVAKLEAALGEAKKQLQDE 74
Cdd:pfam15921 292 SQANSIQSQLEIIQEQARnqnsmymrqLSDLESTVSQLRSELREA---KRMYEDKIEELEKQLVLANSELTEARTERDQF 368
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 75 MLRRVDAENRLQTMKEELDFQKNIYSEELRETKR---RHETRLVEIDNgKQREFESRLADaLQELRAQHEDQVEQYKKEL 151
Cdd:pfam15921 369 SQESGNLDDQLQKLLADLHKREKELSLEKEQNKRlwdRDTGNSITIDH-LRRELDDRNME-VQRLEALLKAMKSECQGQM 446
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 152 EKTYSAkLDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQlsqlQKQLAAKEAKLRDLEDSLARERdtsrRLLAEKER 231
Cdd:pfam15921 447 ERQMAA-IQGKNESLEKVSSLTAQLESTKEMLRKVVEELTAK----KMTLESSERTVSDLTASLQEKE----RAIEATNA 517
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 232 EMAEMRARMQQQLDEYQELldiklaldmeihayrkllEGEEERLRlspsptsqRSRGRASSHSSQTQGGGSVTKKRKLES 311
Cdd:pfam15921 518 EITKLRSRVDLKLQELQHL------------------KNEGDHLR--------NVQTECEALKLQMAEKDKVIEILRQQI 571
|
330 340
....*....|....*....|....*
gi 383792150 312 TESRSSFSQHARTSGRVAVEEVDEE 336
Cdd:pfam15921 572 ENMTQLVGQHGRTAGAMQVEKAQLE 596
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
68-272 |
5.71e-04 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 42.74 E-value: 5.71e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 68 KKQLQDEMLRRVDAENRLQTMKEELDFQKNIYSE------ELRETKRRHETRLVEID------NGKQREFESRLAD--AL 133
Cdd:PRK03918 178 IERLEKFIKRTENIEELIKEKEKELEEVLREINEisselpELREELEKLEKEVKELEelkeeiEELEKELESLEGSkrKL 257
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 134 QELRAQHEDQVEQYKKELEKTYS--AKLDNARQSAERNSNLVgaahEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDL 211
Cdd:PRK03918 258 EEKIRELEERIEELKKEIEELEEkvKELKELKEKAEEYIKLS----EFYEEYLDELREIEKRLSRLEEEINGIEERIKEL 333
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|.
gi 383792150 212 EDSLARERDTSRRlLAEKEREMAEmrarmqqqLDEYQELLDIKLALDMEIHAYRKLLEGEE 272
Cdd:PRK03918 334 EEKEERLEELKKK-LKELEKRLEE--------LEERHELYEEAKAKKEELERLKKRLTGLT 385
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
17-168 |
5.90e-04 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 43.12 E-value: 5.90e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLqdemlrrVDAENRLQTMKEELDFQK 96
Cdd:TIGR02168 870 ELESELEALLNERASLEEALALLRSELEELSEELRELESKRSELRRELEELREKL-------AQLELRLEGLEVRIDNLQ 942
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 383792150 97 NIYSEELRETKRRHETRLVEIDNG--KQREFESRLADALQELRAQHEDQVEQYKKELE-----KTYSAKLDNARQSAER 168
Cdd:TIGR02168 943 ERLSEEYSLTLEEAEALENKIEDDeeEARRRLKRLENKIKELGPVNLAAIEEYEELKErydflTAQKEDLTEAKETLEE 1021
|
|
| PRK09039 |
PRK09039 |
peptidoglycan -binding protein; |
129-237 |
7.12e-04 |
|
peptidoglycan -binding protein;
Pssm-ID: 181619 [Multi-domain] Cd Length: 343 Bit Score: 41.88 E-value: 7.12e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 129 LADALQELRAQhEDQVEQYKKELEKTYSAKLDNARQSAERNSNLVGAAHEELQQS---RIRIDSLSAQLSQLQKQLAAKE 205
Cdd:PRK09039 79 LQDSVANLRAS-LSAAEAERSRLQALLAELAGAGAAAEGRAGELAQELDSEKQVSaraLAQVELLNQQIAALRRQLAALE 157
|
90 100 110
....*....|....*....|....*....|....*....
gi 383792150 206 AKLRDLE----DSLARERDTSRRL---LAEKEREMAEMR 237
Cdd:PRK09039 158 AALDASEkrdrESQAKIADLGRRLnvaLAQRVQELNRYR 196
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
14-250 |
7.50e-04 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 42.62 E-value: 7.50e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 14 DLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELD 93
Cdd:COG1196 561 AAIEYLKAAKAGRATFLPLDKIRARAALAAALARGAIGAAVDLVASDLREADARYYVLGDTLLGRTLVAARLEAALRRAV 640
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 94 fqkniySEELRETKRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEKTYSAKLDNARQSAERNSNLV 173
Cdd:COG1196 641 ------TLAGRLREVTLEGEGGSAGGSLTGGSRRELLAALLEAEAELEELAERLAEEELELEEALLAEEEEERELAEAEE 714
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 174 GAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDtsrrlLAEKEREMAEMRARMQQ-------QLDE 246
Cdd:COG1196 715 ERLEEELEEEALEEQLEAEREELLEELLEEEELLEEEALEELPEPPD-----LEELERELERLEREIEAlgpvnllAIEE 789
|
....
gi 383792150 247 YQEL 250
Cdd:COG1196 790 YEEL 793
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
15-153 |
7.64e-04 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 41.45 E-value: 7.64e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 15 LIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQD--EMLRRVDAENRLQTMKEEL 92
Cdd:COG1579 19 LDRLEHRLKELPAELAELEDELAALEARLEAAKTELEDLEKEIKRLELEIEEVEARIKKyeEQLGNVRNNKEYEALQKEI 98
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....
gi 383792150 93 DFQK---NIYSEELRETKRRHETRLVEIDNGKQRefESRLADALQELRAQHEDQVEQYKKELEK 153
Cdd:COG1579 99 ESLKrriSDLEDEILELMERIEELEEELAELEAE--LAELEAELEEKKAELDEELAELEAELEE 160
|
|
| Surf_Exclu_PgrA |
TIGR04320 |
SEC10/PgrA surface exclusion domain; This model describes a conserved domain found in surface ... |
12-93 |
8.03e-04 |
|
SEC10/PgrA surface exclusion domain; This model describes a conserved domain found in surface proteins of a number of Firmutes. Many members have LPXTG C-terminal anchoring motifs and a substantial number have the KxYKxGKxW putative sorting signal at the N-terminus. The tetracycline resistance plasmid pCF10 in Enterococcus faecalis promotes conjugal plasmid transfer in response to sex pheromones, but PgrA/Sec10 encoded by that plasmid, a member of this family, specifically inhibits the ability of cells to receive homologous plasmids. The phenomenon is called surface exclusion.
Pssm-ID: 275124 [Multi-domain] Cd Length: 356 Bit Score: 42.02 E-value: 8.03e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 12 EGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEA-ALGEAKKQLQDEMLRRVDAENRLQTMKE 90
Cdd:TIGR04320 260 QAKLATAQADLAAAQTALNTAQAALTSAQTAYAAAQAALATAQKELANAQAqALQTAQNNLATAQAALANAEARLAKAKE 339
|
...
gi 383792150 91 ELD 93
Cdd:TIGR04320 340 ALA 342
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
20-276 |
8.04e-04 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 42.36 E-value: 8.04e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 20 ARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKqlqdemLRRVDAENRLQTMKEELDFQKniY 99
Cdd:PRK03918 186 KRTENIEELIKEKEKELEEVLREINEISSELPELREELEKLEKEVKELEE------LKEEIEELEKELESLEGSKRK--L 257
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 100 SEELRETKRRHETRLVEIdngkqREFESRLAD--ALQELRAQHEDQVEQYKKELEKTYSAKLDNARQSAERNSnlVGAAH 177
Cdd:PRK03918 258 EEKIRELEERIEELKKEI-----EELEEKVKElkELKEKAEEYIKLSEFYEEYLDELREIEKRLSRLEEEING--IEERI 330
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 178 EELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARErDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLAl 257
Cdd:PRK03918 331 KELEEKEERLEELKKKLKELEKRLEELEERHELYEEAKAKK-EELERLKKRLTGLTPEKLEKELEELEKAKEEIEEEIS- 408
|
250
....*....|....*....
gi 383792150 258 dmEIHAYRKLLEGEEERLR 276
Cdd:PRK03918 409 --KITARIGELKKEIKELK 425
|
|
| COG1340 |
COG1340 |
Uncharacterized coiled-coil protein, contains DUF342 domain [Function unknown]; |
19-276 |
8.44e-04 |
|
Uncharacterized coiled-coil protein, contains DUF342 domain [Function unknown];
Pssm-ID: 440951 [Multi-domain] Cd Length: 297 Bit Score: 41.43 E-value: 8.44e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 19 QARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLqDEMLRRVdaeNRLQTMKEELDFQKNI 98
Cdd:COG1340 14 EEKIEELREEIEELKEKRDELNEELKELAEKRDELNAQVKELREEAQELREKR-DELNEKV---KELKEERDELNEKLNE 89
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 99 YSEELRETKRRH-ETRLVEID-NGKQREFEsRLADALQ--ELRAQHEDQVEQYKKELEKtysaKLDNARQSAERNSNLVG 174
Cdd:COG1340 90 LREELDELRKELaELNKAGGSiDKLRKEIE-RLEWRQQteVLSPEEEKELVEKIKELEK----ELEKAKKALEKNEKLKE 164
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 175 AAhEELQQSRIRIDSLSAQLsqlqKQLAAKEAKLRDLEDSLARERDTSRrllaEKEREMAEMRARMQQQLDEYQELLDik 254
Cdd:COG1340 165 LR-AELKELRKEAEEIHKKI----KELAEEAQELHEEMIELYKEADELR----KEADELHKEIVEAQEKADELHEEII-- 233
|
250 260
....*....|....*....|..
gi 383792150 255 lALDMEIHAYRKLLEGEEERLR 276
Cdd:COG1340 234 -ELQKELRELRKELKKLRKKQR 254
|
|
| COG0610 |
COG0610 |
Type I site-specific restriction-modification system, R (restriction) subunit and related ... |
52-274 |
8.94e-04 |
|
Type I site-specific restriction-modification system, R (restriction) subunit and related helicases ... [Defense mechanisms];
Pssm-ID: 440375 [Multi-domain] Cd Length: 936 Bit Score: 42.16 E-value: 8.94e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 52 DLRGQVAKLEAALGE-AKKQLQDEMLrrVDAENRLQTMKEELDFQKNIYSEElretkrrhetrlVEIDNGKQREFESRLA 130
Cdd:COG0610 648 DYRGIFENLKKALALySEEDGKEDVL--TDPEEALEELKEALDELRALFPEG------------VDFSAFDPTEKLEALD 713
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 131 DALQELRAQHEDQVEQYK--KELEKTYSAkldnARQSAErnsnLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAK--EA 206
Cdd:COG0610 714 EAVERFLGDEEARKEFKKlfKELSRLYNL----LSPDDE----FGDLELEKYRDDVSFYLALRAKLRKLGEKLDLKeyEE 785
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 383792150 207 KLRDL-EDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEER 274
Cdd:COG0610 786 KIRQLlDEAIDLERKEIKPRIKQNPVQYRKFSELLEEIIEEYNNGALDADEVLEELEELAKEVKEEEER 854
|
|
| mukB |
PRK04863 |
chromosome partition protein MukB; |
57-275 |
8.94e-04 |
|
chromosome partition protein MukB;
Pssm-ID: 235316 [Multi-domain] Cd Length: 1486 Bit Score: 42.25 E-value: 8.94e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 57 VAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELDFQKNIYS---------------EELRETKRRHETRLVEIDNGK 121
Cdd:PRK04863 437 ADNAEDWLEEFQAKEQEATEELLSLEQKLSVAQAAHSQFEQAYQlvrkiagevsrseawDVARELLRRLREQRHLAEQLQ 516
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 122 QREfeSRLADALQELRAQHedQVEQYKKELEKTYSAKLDNA---RQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQ 198
Cdd:PRK04863 517 QLR--MRLSELEQRLRQQQ--RAERLLAEFCKRLGKNLDDEdelEQLQEELEARLESLSESVSEARERRMALRQQLEQLQ 592
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 199 ---KQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEmrarMQQQLDEYQELldiKLALDmEIHAYRKLLEGEEERL 275
Cdd:PRK04863 593 ariQRLAARAPAWLAAQDALARLREQSGEEFEDSQDVTEY----MQQLLEREREL---TVERD-ELAARKQALDEEIERL 664
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
2-275 |
1.01e-03 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 42.08 E-value: 1.01e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 2 GNSEGCNTKKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQD-------E 74
Cdd:pfam01576 401 QDSEHKRKKLEGQLQELQARLSESERQRAELAEKLSKLQSELESVSSLLNEAEGKNIKLSKDVSSLESQLQDtqellqeE 480
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 75 MLRRVDAENRLQTMKEELDFQKNIYSEElrETKRRHETRLVEIDNGKQREFESRLADALQELraqheDQVEQYKKELEKt 154
Cdd:pfam01576 481 TRQKLNLSTRLRQLEDERNSLQEQLEEE--EEAKRNVERQLSTLQAQLSDMKKKLEEDAGTL-----EALEEGKKRLQR- 552
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 155 ysaKLDNARQSAERNSnlvgAAHEELQQSRIR----IDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKE 230
Cdd:pfam01576 553 ---ELEALTQQLEEKA----AAYDKLEKTKNRlqqeLDDLLVDLDHQRQLVSNLEKKQKKFDQMLAEEKAISARYAEERD 625
|
250 260 270 280
....*....|....*....|....*....|....*....|....*
gi 383792150 231 REMAEMRARMQQQLDEYQELLDIKLALDmEIHAYRKLLEGEEERL 275
Cdd:pfam01576 626 RAEAEAREKETRALSLARALEEALEAKE-ELERTNKQLRAEMEDL 669
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
9-276 |
1.05e-03 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 41.93 E-value: 1.05e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 9 TKKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAK------KQLQDEMLRRVDAE 82
Cdd:TIGR04523 148 KKKEKELEKLNNKYNDLKKQKEELENELNLLEKEKLNIQKNIDKIKNKLLKLELLLSNLKkkiqknKSLESQISELKKQN 227
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 83 NRLQTMKEELDFQKNIYSEELRETKRRHETRLVEIDN------GKQREFE------SRLADALQELRAQHEDQVEQYKKE 150
Cdd:TIGR04523 228 NQLKDNIEKKQQEINEKTTEISNTQTQLNQLKDEQNKikkqlsEKQKELEqnnkkiKELEKQLNQLKSEISDLNNQKEQD 307
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 151 LEKTYSAKLDNARQSAERNSNlvgaaheELQQSRIRIDSLSAQLSQLQKQLAAKEAklrdledslarERDTSRRLLAEKE 230
Cdd:TIGR04523 308 WNKELKSELKNQEKKLEEIQN-------QISQNNKIISQLNEQISQLKKELTNSES-----------ENSEKQRELEEKQ 369
|
250 260 270 280
....*....|....*....|....*....|....*....|....*.
gi 383792150 231 REMAEMRARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEERLR 276
Cdd:TIGR04523 370 NEIEKLKKENQSYKQEIKNLESQINDLESKIQNQEKLNQQKDEQIK 415
|
|
| DUF4670 |
pfam15709 |
Domain of unknown function (DUF4670); This family of proteins is found in eukaryotes. Proteins ... |
24-243 |
1.07e-03 |
|
Domain of unknown function (DUF4670); This family of proteins is found in eukaryotes. Proteins in this family are typically between 373 and 763 amino acids in length.
Pssm-ID: 464815 [Multi-domain] Cd Length: 522 Bit Score: 41.86 E-value: 1.07e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 24 DLEALLNSKEAALSTALSEKRTLEGELHD-LRGQVAKLEAALGEAKKQLQDEMLRRVDAE-NRLQTMKEELDFQKNIYSE 101
Cdd:pfam15709 308 NMESEEERSEEDPSKALLEKREQEKASRDrLRAERAEMRRLEVERKRREQEEQRRLQQEQlERAEKMREELELEQQRRFE 387
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 102 ELRETKRRHETrlveiDNGKQREFESRLADALQELRAQHEDQVEQYKKELEKTYSAKLDNARQSAERNSNlvgaaheelq 181
Cdd:pfam15709 388 EIRLRKQRLEE-----ERQRQEEEERKQRLQLQAAQERARQQQEEFRRKLQELQRKKQQEEAERAEAEKQ---------- 452
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....
gi 383792150 182 qsriRIDSLSAQLSQLQKQLA--AKEAKLRDLEDSlaRERDTSRRLLAEKEREMAEMRARMQQQ 243
Cdd:pfam15709 453 ----RQKELEMQLAEEQKRLMemAEEERLEYQRQK--QEAEEKARLEAEERRQKEEEAARLALE 510
|
|
| OmpH |
smart00935 |
Outer membrane protein (OmpH-like); This family includes outer membrane proteins such as OmpH ... |
190-251 |
1.23e-03 |
|
Outer membrane protein (OmpH-like); This family includes outer membrane proteins such as OmpH among others. Skp (OmpH) has been characterized as a molecular chaperone that interacts with unfolded proteins as they emerge in the periplasm from the Sec translocation machinery.
Pssm-ID: 214922 [Multi-domain] Cd Length: 140 Bit Score: 39.49 E-value: 1.23e-03
10 20 30 40 50 60
....*....|....*....|....*....|....*....|....*....|....*....|...
gi 383792150 190 LSAQLSQLQKQLAAKEAKLRDLEDSLARERDT-SRRLLAEKEREMAEMRARMQQQLDEYQELL 251
Cdd:smart00935 23 LEKEFKKRQAELEKLEKELQKLKEKLQKDAATlSEAAREKKEKELQKKVQEFQRKQQKLQQDL 85
|
|
| MukB |
COG3096 |
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell ... |
126-262 |
1.28e-03 |
|
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 442330 [Multi-domain] Cd Length: 1470 Bit Score: 41.86 E-value: 1.28e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 126 ESRLADALQELRAQH--EDQVEQYKKELEKTYSAKLD----NARQSAERNSNLVGAAheELQQSRIRIDSLSAQLSQLQK 199
Cdd:COG3096 518 RAQLAELEQRLRQQQnaERLLEEFCQRIGQQLDAAEEleelLAELEAQLEELEEQAA--EAVEQRSELRQQLEQLRARIK 595
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 200 QLAAKEAKLRDLEDSLARERDTSRRLLAEKeremAEMRARMQQQLD-------EYQELLDIKLALDMEIH 262
Cdd:COG3096 596 ELAARAPAWLAAQDALERLREQSGEALADS----QEVTAAMQQLLErereatvERDELAARKQALESQIE 661
|
|
| SCP-1 |
pfam05483 |
Synaptonemal complex protein 1 (SCP-1); Synaptonemal complex protein 1 (SCP-1) is the major ... |
15-254 |
1.36e-03 |
|
Synaptonemal complex protein 1 (SCP-1); Synaptonemal complex protein 1 (SCP-1) is the major component of the transverse filaments of the synaptonemal complex. Synaptonemal complexes are structures that are formed between homologous chromosomes during meiotic prophase.
Pssm-ID: 114219 [Multi-domain] Cd Length: 787 Bit Score: 41.63 E-value: 1.36e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 15 LIAAQARLKDLEALLNSKEAALSTALsekRTLEGELHDLRGQVAKLEAA-------LGEAKKQLQDEMLRRVDAENRLQT 87
Cdd:pfam05483 420 LLDEKKQFEKIAEELKGKEQELIFLL---QAREKEIHDLEIQLTAIKTSeehylkeVEDLKTELEKEKLKNIELTAHCDK 496
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 88 MKEELDFQKNIYSEELRETKRRHETRlveIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEKT---YSAKLDNARQ 164
Cdd:pfam05483 497 LLLENKELTQEASDMTLELKKHQEDI---INCKKQEERMLKQIENLEEKEMNLRDELESVREEFIQKgdeVKCKLDKSEE 573
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 165 SAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQK---QLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQ 241
Cdd:pfam05483 574 NARSIEYEVLKKEKQMKILENKCNNLKKQIENKNKnieELHQENKALKKKGSAENKQLNAYEIKVNKLELELASAKQKFE 653
|
250
....*....|...
gi 383792150 242 QQLDEYQELLDIK 254
Cdd:pfam05483 654 EIIDNYQKEIEDK 666
|
|
| COG2433 |
COG2433 |
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only]; |
100-276 |
1.48e-03 |
|
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only];
Pssm-ID: 441980 [Multi-domain] Cd Length: 644 Bit Score: 41.38 E-value: 1.48e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 100 SEELRETKRRHETRLVEIDNGKQREfesRLADALQELRAqHEDQVEQYKKELEKTYS------------------AKLDN 161
Cdd:COG2433 312 KEDLSVEEKLHLAREYGYDNDHERD---ALAAALKAYDA-YKNKFERVEKKVPPDVDrdevkarvirglsieealEELIE 387
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 162 ARQSAERNSNLVGAAHEElqqsrIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLlaekEREMAEMRARMQ 241
Cdd:COG2433 388 KELPEEEPEAEREKEHEE-----RELTEEEEEIRRLEEQVERLEAEVEELEAELEEKDERIERL----ERELSEARSEER 458
|
170 180 190
....*....|....*....|....*....|....*
gi 383792150 242 QQLDEYQELldikLALDMEIHAYRKLLEGEEERLR 276
Cdd:COG2433 459 REIRKDREI----SRLDREIERLERELEEERERIE 489
|
|
| OmpH |
pfam03938 |
Outer membrane protein (OmpH-like); This family includes outer membrane proteins such as OmpH ... |
190-251 |
1.49e-03 |
|
Outer membrane protein (OmpH-like); This family includes outer membrane proteins such as OmpH among others. Skp (OmpH) has been characterized as a molecular chaperone that interacts with unfolded proteins as they emerge in the periplasm from the Sec translocation machinery.
Pssm-ID: 461098 [Multi-domain] Cd Length: 140 Bit Score: 39.10 E-value: 1.49e-03
10 20 30 40 50 60
....*....|....*....|....*....|....*....|....*....|....*....|..
gi 383792150 190 LSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELL 251
Cdd:pfam03938 24 LEKKFKKRQAELEAKQKELQKLYEELQKDGALLEEEREEKEQELQKKEQELQQLQQKAQQEL 85
|
|
| PRK09039 |
PRK09039 |
peptidoglycan -binding protein; |
186-252 |
1.62e-03 |
|
peptidoglycan -binding protein;
Pssm-ID: 181619 [Multi-domain] Cd Length: 343 Bit Score: 40.72 E-value: 1.62e-03
10 20 30 40 50 60 70
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 383792150 186 RIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKER------EMAEMRARMQQQLDEYQELLD 252
Cdd:PRK09039 54 ALDRLNSQIAELADLLSLERQGNQDLQDSVANLRASLSAAEAERSRlqallaELAGAGAAAEGRAGELAQELD 126
|
|
| DUF3584 |
pfam12128 |
Protein of unknown function (DUF3584); This protein is found in bacteria and eukaryotes. ... |
10-240 |
1.62e-03 |
|
Protein of unknown function (DUF3584); This protein is found in bacteria and eukaryotes. Proteins in this family are typically between 943 to 1234 amino acids in length. This family contains a P-loop motif suggesting it is a nucleotide binding protein. It may be involved in replication.
Pssm-ID: 432349 [Multi-domain] Cd Length: 1191 Bit Score: 41.36 E-value: 1.62e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 10 KKEGDLIAAQARLKDLE---ALLNSKEAALSTALSE-KRTLEGELHDL-------RGQVAKLEAALGEAKKQLQDEmLRR 78
Cdd:pfam12128 608 KAEEALQSAREKQAAAEeqlVQANGELEKASREETFaRTALKNARLDLrrlfdekQSEKDKKNKALAERKDSANER-LNS 686
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 79 VDAEnrlqtmKEELDFQKNIYSEELRETKRRHET------RLVEIDNGKQREFESRLADALQELRAQHEDQVE-QYKKEL 151
Cdd:pfam12128 687 LEAQ------LKQLDKKHQAWLEEQKEQKREARTekqaywQVVEGALDAQLALLKAAIAARRSGAKAELKALEtWYKRDL 760
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 152 E----------------KTYSAKLDNARQ-SAERNSNLVGAAHEELQQS---RIRIDSLSAQLSQLQKQLAAKEAKLRDL 211
Cdd:pfam12128 761 AslgvdpdviaklkreiRTLERKIERIAVrRQEVLRYFDWYQETWLQRRprlATQLSNIERAISELQQQLARLIADTKLR 840
|
250 260
....*....|....*....|....*....
gi 383792150 212 EDSLARERDTSRRLLAEKEREMAEMRARM 240
Cdd:pfam12128 841 RAKLEMERKASEKQQVRLSENLRGLRCEM 869
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
14-227 |
2.13e-03 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 41.08 E-value: 2.13e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 14 DLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELD 93
Cdd:COG1196 631 RLEAALRRAVTLAGRLREVTLEGEGGSAGGSLTGGSRRELLAALLEAEAELEELAERLAEEELELEEALLAEEEEERELA 710
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 94 FQKNIYSEELRETKRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQykkELEKtysaKLDNARQSAER--NSN 171
Cdd:COG1196 711 EAEEERLEEELEEEALEEQLEAEREELLEELLEEEELLEEEALEELPEPPDLE---ELER----ELERLEREIEAlgPVN 783
|
170 180 190 200 210
....*....|....*....|....*....|....*....|....*....|....*.
gi 383792150 172 LvgAAHEELQQSRIRIDSLSAQLSQLQKqlaAKEaKLRDLEDSLarERDTSRRLLA 227
Cdd:COG1196 784 L--LAIEEYEELEERYDFLSEQREDLEE---ARE-TLEEAIEEI--DRETRERFLE 831
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
128-277 |
2.44e-03 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 40.91 E-value: 2.44e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 128 RLADALQELRAQHEdQVEQYKKELEKtYSAKLDNARQsAERNSNLVGAAHEELQQSRIRIDSLSAQ---LSQLQKQLAAK 204
Cdd:COG4717 92 ELQEELEELEEELE-ELEAELEELRE-ELEKLEKLLQ-LLPLYQELEALEAELAELPERLEELEERleeLRELEEELEEL 168
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 383792150 205 EAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDiklALDMEIHAYRKLLEGEEERLRL 277
Cdd:COG4717 169 EAELAELQEELEELLEQLSLATEEELQDLAEELEELQQRLAELEEELE---EAQEELEELEEELEQLENELEA 238
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
19-298 |
2.69e-03 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 40.93 E-value: 2.69e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 19 QARLKDLEALLNSKEAALSTALSEKRTLEGELHDLR-------GQVAKLEAALGEAKKQLQ-------DEMLRRVDAENR 84
Cdd:pfam01576 186 EAMISDLEERLKKEEKGRQELEKAKRKLEGESTDLQeqiaelqAQIAELRAQLAKKEEELQaalarleEETAQKNNALKK 265
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 85 LQTMKEEL-DFQKNIYSEELRETKRRHETR-LVEIDNGKQREFESRL--ADALQELRAQHEDQVEQYKKELE---KTYSA 157
Cdd:pfam01576 266 IRELEAQIsELQEDLESERAARNKAEKQRRdLGEELEALKTELEDTLdtTAAQQELRSKREQEVTELKKALEeetRSHEA 345
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 158 KLDNARQsaeRNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLlaekEREMAEMR 237
Cdd:pfam01576 346 QLQEMRQ---KHTQALEELTEQLEQAKRNKANLEKAKQALESENAELQAELRTLQQAKQDSEHKRKKL----EGQLQELQ 418
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|.
gi 383792150 238 ARMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEERlrlspsptSQRSRGRASSHSSQTQ 298
Cdd:pfam01576 419 ARLSESERQRAELAEKLSKLQSELESVSSLLNEAEGK--------NIKLSKDVSSLESQLQ 471
|
|
| OmpH |
smart00935 |
Outer membrane protein (OmpH-like); This family includes outer membrane proteins such as OmpH ... |
58-151 |
3.12e-03 |
|
Outer membrane protein (OmpH-like); This family includes outer membrane proteins such as OmpH among others. Skp (OmpH) has been characterized as a molecular chaperone that interacts with unfolded proteins as they emerge in the periplasm from the Sec translocation machinery.
Pssm-ID: 214922 [Multi-domain] Cd Length: 140 Bit Score: 38.33 E-value: 3.12e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 58 AKLEAALGEAKKQLQDEmlrrvdaENRLQTMKEELDFQKNIYSEELRETKRRHETRLVEIDNGKQREFESRLADALQELR 137
Cdd:smart00935 21 KQLEKEFKKRQAELEKL-------EKELQKLKEKLQKDAATLSEAAREKKEKELQKKVQEFQRKQQKLQQDLQKRQQEEL 93
|
90
....*....|....
gi 383792150 138 AQHEDQVEQYKKEL 151
Cdd:smart00935 94 QKILDKINKAIKEV 107
|
|
| LCD1 |
pfam09798 |
DNA damage checkpoint protein; This is a family of proteins which regulate checkpoint kinases. ... |
190-316 |
3.62e-03 |
|
DNA damage checkpoint protein; This is a family of proteins which regulate checkpoint kinases. In Schizosaccharomyces pombe this protein is called Rad26 and in Saccharomyces cerevisiae it is called LCD1.
Pssm-ID: 462906 Cd Length: 615 Bit Score: 40.38 E-value: 3.62e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 190 LSAQLSQLQKQLAAKEAKLRDLEDSLARerdtsrrllaEKEREMAEMRArmqqqldEYQELLDIKLALDMEIhayRKLle 269
Cdd:pfam09798 2 LRDKLELLQQEKEKELEKLKNSYEELKS----------SHEEELEKLKQ-------EVQKLEDEKKFLLNEL---RSL-- 59
|
90 100 110 120
....*....|....*....|....*....|....*....|....*..
gi 383792150 270 geeerlrLSPSPTSQRSRGRASSHSSQtqgggSVTKKRKLESTESRS 316
Cdd:pfam09798 60 -------SATSPASSQSHETDTDDSSS-----VSLKKRKIEESTAES 94
|
|
| PRK10929 |
PRK10929 |
putative mechanosensitive channel protein; Provisional |
145-313 |
4.07e-03 |
|
putative mechanosensitive channel protein; Provisional
Pssm-ID: 236798 [Multi-domain] Cd Length: 1109 Bit Score: 40.04 E-value: 4.07e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 145 EQYKKELEKTYSAKLDN------ARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARE 218
Cdd:PRK10929 26 KQITQELEQAKAAKTPAqaeiveALQSALNWLEERKGSLERAKQYQQVIDNFPKLSAELRQQLNNERDEPRSVPPNMSTD 105
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 219 R------DTSRRLLaEKEREmaemrarMQQQLDEYQELLDIKLALDMEIHAYRKLLEGEEERLRLSPSPTSqrSRGRASS 292
Cdd:PRK10929 106 AleqeilQVSSQLL-EKSRQ-------AQQEQDRAREISDSLSQLPQQQTEARRQLNEIERRLQTLGTPNT--PLAQAQL 175
|
170 180
....*....|....*....|.
gi 383792150 293 HSSQTQgggSVTKKRKLESTE 313
Cdd:PRK10929 176 TALQAE---SAALKALVDELE 193
|
|
| Apolipoprotein |
pfam01442 |
Apolipoprotein A1/A4/E domain; These proteins contain several 22 residue repeats which form a ... |
80-252 |
4.16e-03 |
|
Apolipoprotein A1/A4/E domain; These proteins contain several 22 residue repeats which form a pair of alpha helices. This family includes: Apolipoprotein A-I. Apolipoprotein A-IV. Apolipoprotein E.
Pssm-ID: 460211 [Multi-domain] Cd Length: 175 Bit Score: 38.40 E-value: 4.16e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 80 DAENRLQTMKEELDFQKNIYSEELRETKRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEkTYSAKL 159
Cdd:pfam01442 4 DSLDELSTYAEELQEQLGPVAQELVDRLEKETEALRERLQKDLEEVRAKLEPYLEELQAKLGQNVEELRQRLE-PYTEEL 82
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 160 -DNARQSAERNSNLVGAAHEELQQS-RIRIDSLSAQLSQLQKQLAAK-EAKLRDLEDSLARERDTSRRLLAEKERemaEM 236
Cdd:pfam01442 83 rKRLNADAEELQEKLAPYGEELRERlEQNVDALRARLAPYAEELRQKlAERLEELKESLAPYAEEVQAQLSQRLQ---EL 159
|
170
....*....|....*.
gi 383792150 237 RARMQQQLDEYQELLD 252
Cdd:pfam01442 160 REKLEPQAEDLREKLD 175
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
12-243 |
4.23e-03 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 40.02 E-value: 4.23e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 12 EGDLIAAQARLKDLEALLNS----------KEAALSTALSEKR----TLEGELHDLRGQVAKLEA------ALGEAKKQL 71
Cdd:PRK02224 432 EATLRTARERVEEAEALLEAgkcpecgqpvEGSPHVETIEEDRerveELEAELEDLEEEVEEVEErleraeDLVEAEDRI 511
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 72 QDEMLRRVDAENRLQTMKEELDfQKNIYSEELRETKRRHETRLVEID--------------------NGKQREFESRL-- 129
Cdd:PRK02224 512 ERLEERREDLEELIAERRETIE-EKRERAEELRERAAELEAEAEEKReaaaeaeeeaeeareevaelNSKLAELKERIes 590
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 130 --------------ADALQELRAQHEDQVEQYKKELEKtYSAKLDNARQSAERnsnLVGAAHEELQQSRIR--------- 186
Cdd:PRK02224 591 lerirtllaaiadaEDEIERLREKREALAELNDERRER-LAEKRERKRELEAE---FDEARIEEAREDKERaeeyleqve 666
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 383792150 187 --IDSLSAQLSQLQKQLAAKEAKLRDLEdSLARERDT--SRRLLAEKEREMAE--------MRARMQQQ 243
Cdd:PRK02224 667 ekLDELREERDDLQAEIGAVENELEELE-ELRERREAleNRVEALEALYDEAEelesmygdLRAELRQR 734
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
179-275 |
4.38e-03 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 39.14 E-value: 4.38e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 179 ELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLAR---ERDTSRRLLAEKEREMAEMRARM---QQQLD------E 246
Cdd:COG1579 11 DLQELDSELDRLEHRLKELPAELAELEDELAALEARLEAaktELEDLEKEIKRLELEIEEVEARIkkyEEQLGnvrnnkE 90
|
90 100 110 120
....*....|....*....|....*....|....*....|...
gi 383792150 247 YQELL--------------DIKLALDMEIHAYRKLLEGEEERL 275
Cdd:COG1579 91 YEALQkeieslkrrisdleDEILELMERIEELEEELAELEAEL 133
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
14-252 |
4.43e-03 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 40.00 E-value: 4.43e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 14 DLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEE-L 92
Cdd:TIGR04523 104 DLSKINSEIKNDKEQKNKLEVELNKLEKQKKENKKNIDKFLTEIKKKEKELEKLNNKYNDLKKQKEELENELNLLEKEkL 183
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 93 DFQKNIysEELRETKRRHETRLVEID--NGKQREFESRLADaLQELRAQHEDQVEQYKKELEKTySAKLDNARQSAERNS 170
Cdd:TIGR04523 184 NIQKNI--DKIKNKLLKLELLLSNLKkkIQKNKSLESQISE-LKKQNNQLKDNIEKKQQEINEK-TTEISNTQTQLNQLK 259
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 171 NLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSlaRERDTSRRL---LAEKEREMAEmrarMQQQLDEY 247
Cdd:TIGR04523 260 DEQNKIKKQLSEKQKELEQNNKKIKELEKQLNQLKSEISDLNNQ--KEQDWNKELkseLKNQEKKLEE----IQNQISQN 333
|
....*
gi 383792150 248 QELLD 252
Cdd:TIGR04523 334 NKIIS 338
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
5-252 |
5.25e-03 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 39.77 E-value: 5.25e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 5 EGCNTKKEGDLIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENR 84
Cdd:pfam01576 116 EAARQKLQLEKVTTEAKIKKLEEDILLLEDQNSKLSKERKLLEERISEFTSNLAEEEEKAKSLSKLKNKHEAMISDLEER 195
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 85 LQtmKEELDFQkniyseELRETKRRHETRLVEIdngkqREFESRLADALQELRAQ---HEDQVEQYKKELEKTYSAKlDN 161
Cdd:pfam01576 196 LK--KEEKGRQ------ELEKAKRKLEGESTDL-----QEQIAELQAQIAELRAQlakKEEELQAALARLEEETAQK-NN 261
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 162 ARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLA--------RERDTS--RRLLAEK-- 229
Cdd:pfam01576 262 ALKKIRELEAQISELQEDLESERAARNKAEKQRRDLGEELEALKTELEDTLDTTAaqqelrskREQEVTelKKALEEEtr 341
|
250 260
....*....|....*....|....*
gi 383792150 230 --EREMAEMRARMQQQLDEYQELLD 252
Cdd:pfam01576 342 shEAQLQEMRQKHTQALEELTEQLE 366
|
|
| PTZ00121 |
PTZ00121 |
MAEBL; Provisional |
17-249 |
5.32e-03 |
|
MAEBL; Provisional
Pssm-ID: 173412 [Multi-domain] Cd Length: 2084 Bit Score: 40.12 E-value: 5.32e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 17 AAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELDFQK 96
Cdd:PTZ00121 1566 AEEAKKAEEDKNMALRKAEEAKKAEEARIEEVMKLYEEEKKMKAEEAKKAEEAKIKAEELKKAEEEKKKVEQLKKKEAEE 1645
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 97 NIYSEELRETKRRHETRLVEIdnGKQREFESRLADALQELRAQHEDQVEQYKKELEKtySAKLDNARQSAERNSNLVGAA 176
Cdd:PTZ00121 1646 KKKAEELKKAEEENKIKAAEE--AKKAEEDKKKAEEAKKAEEDEKKAAEALKKEAEE--AKKAEELKKKEAEEKKKAEEL 1721
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|...
gi 383792150 177 HEELQQSRIRIDSLSAQLSQLQKQlaAKEAKLRDLEDSLARERDTSRRLLAEKEREmaEMRARMQQQLDEYQE 249
Cdd:PTZ00121 1722 KKAEEENKIKAEEAKKEAEEDKKK--AEEAKKDEEEKKKIAHLKKEEEKKAEEIRK--EKEAVIEEELDEEDE 1790
|
|
| mukB |
PRK04863 |
chromosome partition protein MukB; |
16-256 |
5.64e-03 |
|
chromosome partition protein MukB;
Pssm-ID: 235316 [Multi-domain] Cd Length: 1486 Bit Score: 39.94 E-value: 5.64e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 16 IAAQARLKDLEALlnSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDemlrrvdAENRLQTMKEELDFQ 95
Cdd:PRK04863 277 HANERRVHLEEAL--ELRRELYTSRRQLAAEQYRLVEMARELAELNEAESDLEQDYQA-------ASDHLNLVQTALRQQ 347
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 96 KNI--YSEELRETKRRHE--TRLVEIDNGKQREFESRLADALQE---LRAQHEDQVEQYkkELEKTYSAKLDNARQSAER 168
Cdd:PRK04863 348 EKIerYQADLEELEERLEeqNEVVEEADEQQEENEARAEAAEEEvdeLKSQLADYQQAL--DVQQTRAIQYQQAVQALER 425
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 169 NSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDslARERDTS-----RRLLAEKEREMA-------EM 236
Cdd:PRK04863 426 AKQLCGLPDLTADNAEDWLEEFQAKEQEATEELLSLEQKLSVAQA--AHSQFEQayqlvRKIAGEVSRSEAwdvarelLR 503
|
250 260
....*....|....*....|
gi 383792150 237 RARMQQQLDEYQELLDIKLA 256
Cdd:PRK04863 504 RLREQRHLAEQLQQLRMRLS 523
|
|
| COG4372 |
COG4372 |
Uncharacterized protein, contains DUF3084 domain [Function unknown]; |
15-322 |
5.91e-03 |
|
Uncharacterized protein, contains DUF3084 domain [Function unknown];
Pssm-ID: 443500 [Multi-domain] Cd Length: 370 Bit Score: 39.12 E-value: 5.91e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 15 LIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMKEELD- 93
Cdd:COG4372 33 LRKALFELDKLQEELEQLREELEQAREELEQLEEELEQARSELEQLEEELEELNEQLQAAQAELAQAQEELESLQEEAEe 112
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 94 -------FQKNIYSEELRETKRRHETRLVEIDNGKQREFESRLADALQELRAQHEDQVEQYKKELEKTYSAKLDNARQSA 166
Cdd:COG4372 113 lqeeleeLQKERQDLEQQRKQLEAQIAELQSEIAEREEELKELEEQLESLQEELAALEQELQALSEAEAEQALDELLKEA 192
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 167 ERNSnLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARMQQQLDE 246
Cdd:COG4372 193 NRNA-EKEEELAEAEKLIESLPRELAEELLEAKDSLEAKLGLALSALLDALELEEDKEELLEEVILKEIEELELAILVEK 271
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 383792150 247 YQELLDIKLALDMEIHAYRKLLEGEEERLRLSPSPTSQRSRGRASSHSSQTQGGGSVTKKRKLESTESRSSFSQHA 322
Cdd:COG4372 272 DTEEEELEIAALELEALEEAALELKLLALLLNLAALSLIGALEDALLAALLELAKKLELALAILLAELADLLQLLL 347
|
|
| PRK09039 |
PRK09039 |
peptidoglycan -binding protein; |
189-274 |
5.94e-03 |
|
peptidoglycan -binding protein;
Pssm-ID: 181619 [Multi-domain] Cd Length: 343 Bit Score: 39.18 E-value: 5.94e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 189 SLSAQLSQLQKQLAAKEA---KLRDLEDSLARERDTSRRLLAEKEREMAEMR---ARMQQQLdeyqELLDIKL-ALDMEI 261
Cdd:PRK09039 78 DLQDSVANLRASLSAAEAersRLQALLAELAGAGAAAEGRAGELAQELDSEKqvsARALAQV----ELLNQQIaALRRQL 153
|
90
....*....|...
gi 383792150 262 HAYRKLLEGEEER 274
Cdd:PRK09039 154 AALEAALDASEKR 166
|
|
| PRK12704 |
PRK12704 |
phosphodiesterase; Provisional |
8-153 |
6.57e-03 |
|
phosphodiesterase; Provisional
Pssm-ID: 237177 [Multi-domain] Cd Length: 520 Bit Score: 39.38 E-value: 6.57e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 8 NTKKEGDLIAAQAR-----LKDlEALLNSKEAALStalsEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAE 82
Cdd:PRK12704 35 EAEEEAKRILEEAKkeaeaIKK-EALLEAKEEIHK----LRNEFEKELRERRNELQKLEKRLLQKEENLDRKLELLEKRE 109
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 83 NRLQTMKEELDFQKNIY---SEELRETKRRHETRLVEIDNGKQREFESRLADALQElRAQHEDQV------EQYKKELEK 153
Cdd:PRK12704 110 EELEKKEKELEQKQQELekkEEELEELIEEQLQELERISGLTAEEAKEILLEKVEE-EARHEAAVlikeieEEAKEEADK 188
|
|
| sbcc |
TIGR00618 |
exonuclease SbcC; All proteins in this family for which functions are known are part of an ... |
15-277 |
6.94e-03 |
|
exonuclease SbcC; All proteins in this family for which functions are known are part of an exonuclease complex with sbcD homologs. This complex is involved in the initiation of recombination to regulate the levels of palindromic sequences in DNA. This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University). [DNA metabolism, DNA replication, recombination, and repair]
Pssm-ID: 129705 [Multi-domain] Cd Length: 1042 Bit Score: 39.57 E-value: 6.94e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 15 LIAAQARLKDLEALLNSKEAALSTALSEKRTLEGELHDLRGQVAKLEAALGEAKKQLQDEMLRRVDAENRLQTMK----- 89
Cdd:TIGR00618 381 IHTLQQQKTTLTQKLQSLCKELDILQREQATIDTRTSAFRDLQGQLAHAKKQQELQQRYAELCAAAITCTAQCEKlekih 460
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 90 -----------EELDFQKNIYSEELRETKRRHETRLVEIdNGKQREFESRLADALQELRA------------QHEDQVEQ 146
Cdd:TIGR00618 461 lqesaqslkerEQQLQTKEQIHLQETRKKAVVLARLLEL-QEEPCPLCGSCIHPNPARQDidnpgpltrrmqRGEQTYAQ 539
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 147 YKKELEKT-------------YSAKLDNARQS--------------------------------AERNSNLVGAAHEELQ 181
Cdd:TIGR00618 540 LETSEEDVyhqltserkqrasLKEQMQEIQQSfsiltqcdnrskedipnlqnitvrlqdlteklSEAEDMLACEQHALLR 619
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 182 QSRIRID--SLSAQLSQLQKQLAAKEAKLRDLEDSLA--RERDTSRRLLAEKEREMAEMRARMQQQLDEYQELLDIKLAL 257
Cdd:TIGR00618 620 KLQPEQDlqDVRLHLQQCSQELALKLTALHALQLTLTqeRVREHALSIRVLPKELLASRQLALQKMQSEKEQLTYWKEML 699
|
330 340
....*....|....*....|
gi 383792150 258 DMEIHAYRKLLEGEEERLRL 277
Cdd:TIGR00618 700 AQCQTLLRELETHIEEYDRE 719
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
81-275 |
7.19e-03 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 39.28 E-value: 7.19e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 81 AENRLQTMKEELDFQKNIYSEELRETKrrhetrlvEIDNgKQREFESRLADALQELRaQHEDQVEQYKKELEKtysakld 160
Cdd:PRK03918 163 AYKNLGEVIKEIKRRIERLEKFIKRTE--------NIEE-LIKEKEKELEEVLREIN-EISSELPELREELEK------- 225
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 161 narqsaernsnlVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRlLAEKEREMAEMRARM 240
Cdd:PRK03918 226 ------------LEKEVKELEELKEEIEELEKELESLEGSKRKLEEKIRELEERIEELKKEIEE-LEEKVKELKELKEKA 292
|
170 180 190
....*....|....*....|....*....|....*...
gi 383792150 241 QQQL---DEYQELLDIKLALDMEIHAYRKLLEGEEERL 275
Cdd:PRK03918 293 EEYIklsEFYEEYLDELREIEKRLSRLEEEINGIEERI 330
|
|
| MukB |
COG3096 |
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell ... |
45-196 |
7.43e-03 |
|
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 442330 [Multi-domain] Cd Length: 1470 Bit Score: 39.55 E-value: 7.43e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 45 TLEGELHDLRGQVAKLEAALgEAKKQLQDEMLRRVDAENRLQTMKEELDFQKNIYSEELRE-TKRRHETR--LVEIdNGK 121
Cdd:COG3096 516 QLRAQLAELEQRLRQQQNAE-RLLEEFCQRIGQQLDAAEELEELLAELEAQLEELEEQAAEaVEQRSELRqqLEQL-RAR 593
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 122 QREFESR------LADALQELRAQHEDQVEQykkelektySAKLDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLS 195
Cdd:COG3096 594 IKELAARapawlaAQDALERLREQSGEALAD---------SQEVTAAMQQLLEREREATVERDELAARKQALESQIERLS 664
|
.
gi 383792150 196 Q 196
Cdd:COG3096 665 Q 665
|
|
| CusB_dom_1 |
pfam00529 |
Cation efflux system protein CusB domain 1; The cation efflux system protein CusB from E. coli ... |
141-298 |
8.89e-03 |
|
Cation efflux system protein CusB domain 1; The cation efflux system protein CusB from E. coli can be divided into four different domains, the first three domains of the protein are mostly beta-strands and the fourth forms an all alpha-helical domain. This entry represents the first beta-domain (domain 1) of CusB and it is formed by the N and C-terminal ends of the polypeptide (residues 89-102 and 324-385). CusB is part of the copper-transporting efflux system CusCFBA. This domain can also be found in other membrane-fusion proteins, such as HlyD, MdtN, MdtE and AaeA. HlyD is a component of the prototypical alpha-haemolysin (HlyA) bacterial type I secretion system, along with the other components HlyB and TolC. HlyD is anchored in the cytoplasmic membrane by a single transmembrane domain and has a large periplasmic domain within the carboxy-terminal 100 amino acids, HlyB and HlyD form a stable complex that binds the recombinant protein bearing a C-terminal HlyA signal sequence and ATP in the cytoplasm. HlyD, HlyB and TolC combine to form the three-component ABC transporter complex that forms a trans-membrane channel or pore through which HlyA can be transferred directly to the extracellular medium. Cutinase has been shown to be transported effectively through this pore.
Pssm-ID: 425733 [Multi-domain] Cd Length: 322 Bit Score: 38.56 E-value: 8.89e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 141 EDQVEQYKKELektysAKLDNARQSAERNSNLVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARerd 220
Cdd:pfam00529 57 QAALDSAEAQL-----AKAQAQVARLQAELDRLQALESELAISRQDYDGATAQLRAAQAAVKAAQAQLAQAQIDLAR--- 128
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 221 tsRRLLAEK----EREMAEMRARMQQQLDEY----QELLDIKLALDMEIHAYRKLLEGEEERLRLSPSpTSQRSRGRASS 292
Cdd:pfam00529 129 --RRVLAPIggisRESLVTAGALVAQAQANLlatvAQLDQIYVQITQSAAENQAEVRSELSGAQLQIA-EAEAELKLAKL 205
|
....*.
gi 383792150 293 HSSQTQ 298
Cdd:pfam00529 206 DLERTE 211
|
|
| PRK04778 |
PRK04778 |
septation ring formation regulator EzrA; Provisional |
46-201 |
9.28e-03 |
|
septation ring formation regulator EzrA; Provisional
Pssm-ID: 179877 [Multi-domain] Cd Length: 569 Bit Score: 38.66 E-value: 9.28e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 46 LEGELHDLRGQVAKLEAALG-----EAKKQLQD---------EMLRR-VDAEN----RLQTMKEELDFQKNIYSEELRET 106
Cdd:PRK04778 254 IEKEIQDLKEQIDENLALLEeldldEAEEKNEEiqeridqlyDILEReVKARKyvekNSDTLPDFLEHAKEQNKELKEEI 333
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 383792150 107 KRRHET-RLVEIDNGKQREFESRladaLQELRAQHEDQVEQYKKElEKTYSAKLDNArQSAERNSNLVGAAHEELQQS-- 183
Cdd:PRK04778 334 DRVKQSyTLNESELESVRQLEKQ----LESLEKQYDEITERIAEQ-EIAYSELQEEL-EEILKQLEEIEKEQEKLSEMlq 407
|
170 180
....*....|....*....|
gi 383792150 184 RIRIDSLSAQ--LSQLQKQL 201
Cdd:PRK04778 408 GLRKDELEARekLERYRNKL 427
|
|
| PRK10920 |
PRK10920 |
putative uroporphyrinogen III C-methyltransferase; Provisional |
172-240 |
9.33e-03 |
|
putative uroporphyrinogen III C-methyltransferase; Provisional
Pssm-ID: 236795 Cd Length: 390 Bit Score: 38.54 E-value: 9.33e-03
10 20 30 40 50 60
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 383792150 172 LVGAAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLEDSLARERDTSRRLLAEKEREMAEMRARM 240
Cdd:PRK10920 54 LYYHGKQQAQNQTATNDALANQLTALQKAQESQKQELEGILKQQAKALDQANRQQAALAKQLDELQQKV 122
|
|
| AcrA |
COG0845 |
Multidrug efflux pump subunit AcrA (membrane-fusion protein) [Cell wall/membrane/envelope ... |
141-212 |
9.89e-03 |
|
Multidrug efflux pump subunit AcrA (membrane-fusion protein) [Cell wall/membrane/envelope biogenesis, Defense mechanisms];
Pssm-ID: 440606 [Multi-domain] Cd Length: 324 Bit Score: 38.39 E-value: 9.89e-03
10 20 30 40 50 60 70
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 383792150 141 EDQVEQYKKELEKTYsAKLDNARQSAERNSNLVG---AAHEELQQSRIRIDSLSAQLSQLQKQLAAKEAKLRDLE 212
Cdd:COG0845 60 QAALAQAQAQLAAAQ-AQLELAKAELERYKALLKkgaVSQQELDQAKAALDQAQAALAAAQAALEQARANLAYTT 133
|
|
| |