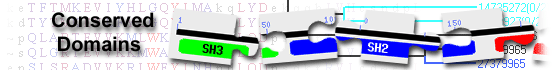|
Name |
Accession |
Description |
Interval |
E-value |
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
18-491 |
7.11e-18 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 88.97 E-value: 7.11e-18
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 18 QKIQELTTNAKETHTKLALAEARVQEEEQKATRLEkELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALS---RQI 94
Cdd:PRK03918 207 REINEISSELPELREELEKLEKEVKELEELKEEIE-ELEKELESLEGSKRKLEEKIRELEERIEELKKEIEELEekvKEL 285
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 95 DELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLER--------- 165
Cdd:PRK03918 286 KELKEKAEEYIKLSEFYEEYLDELREIEKRLSRLEEEINGIEERIKELEEKEERLEELKKKLKELEKRLEEleerhelye 365
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 166 ---------ETLQSKDFKLEVEKLSKRIMALEK----LEDAFNKSKQECYSLKcNLEKERMTT----------------- 215
Cdd:PRK03918 366 eakakkeelERLKKRLTGLTPEKLEKELEELEKakeeIEEEISKITARIGELK-KEIKELKKAieelkkakgkcpvcgre 444
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 216 ----------KQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVM--FVDERKTMSEKLKKTE-DKLQAAS 282
Cdd:PRK03918 445 lteehrkellEEYTAELKRIEKELKEIEEKERKLRKELRELEKVLKKESELIKLkeLAEQLKELEEKLKKYNlEELEKKA 524
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 283 SQLQVEQNKVTTVT------EKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLkaeEEKGndlLSRVNMLKNRLQSLE 356
Cdd:PRK03918 525 EEYEKLKEKLIKLKgeikslKKELEKLEELKKKLAELEKKLDELEEELAELLKEL---EELG---FESVEELEERLKELE 598
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 357 AIEKDFLKNK--------LNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAI--EDDLMKTEDEYETLERRYANE 426
Cdd:PRK03918 599 PFYNEYLELKdaekelerEEKELKKLEEELDKAFEELAETEKRLEELRKELEELEKKysEEEYEELREEYLELSRELAGL 678
|
490 500 510 520 530 540
....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 544346182 427 RDKAQFLSKELEHVKMELAKYKlAEKTEtsheqwlFKRLQEEEAKSGHLSREVDALKEKIHEYMA 491
Cdd:PRK03918 679 RAELEELEKRREEIKKTLEKLK-EELEE-------REKAKKELEKLEKALERVEELREKVKKYKA 735
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
3-533 |
1.02e-17 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 88.58 E-value: 1.02e-17
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 3 VDEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTK-----------------FHQD 65
Cdd:TIGR02168 238 REELEELQEELKEAEEELEELTAELQELEEKLEELRLEVSELEEEIEELQKELYALANEisrleqqkqilrerlanLERQ 317
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 66 QDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKIskgEYGNAGIMAEVEELRKRVLDMEGK 145
Cdd:TIGR02168 318 LEELEAQLEELESKLDELAEELAELEEKLEELKEELESLEAELEELEAELEEL---ESRLEELEEQLETLRSKVAQLELQ 394
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 146 ----DEELIKMEEQCRDLNKRLERET---------LQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKER 212
Cdd:TIGR02168 395 iaslNNEIERLEARLERLEDRRERLQqeieellkkLEEAELKELQAELEELEEELEELQEELERLEEALEELREELEEAE 474
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 213 MTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMF--------VDE--RKTMSEKL---------KK 273
Cdd:TIGR02168 475 QALDAAERELAQLQARLDSLERLQENLEGFSEGVKALLKNQSGLSGILgvlselisVDEgyEAAIEAALggrlqavvvEN 554
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 274 TEDKLQAASSQLQVEQNKVTTVTEKLIEETKrALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQ 353
Cdd:TIGR02168 555 LNAAKKAIAFLKQNELGRVTFLPLDSIKGTE-IQGNDREILKNIEGFLGVAKDLVKFDPKLRKALSYLLGGVLVVDDLDN 633
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 354 SLE-AIEKDFLKNKLNQD--------------SGKSTTALHQENN-------------KIKELSQEVERLKLKLKDMKAI 405
Cdd:TIGR02168 634 ALElAKKLRPGYRIVTLDgdlvrpggvitggsAKTNSSILERRREieeleekieeleeKIAELEKALAELRKELEELEEE 713
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 406 EDDLMKTEDEYET----LERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDA 481
Cdd:TIGR02168 714 LEQLRKELEELSRqisaLRKDLARLEAEVEQLEERIAQLSKELTELEAEIEELEERLEEAEEELAEAEAEIEELEAQIEQ 793
|
570 580 590 600 610
....*....|....*....|....*....|....*....|....*....|..
gi 544346182 482 LKEkihEYMATEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKELER 533
Cdd:TIGR02168 794 LKE---ELKALREALDELRAELTLLNEEAANLRERLESLERRIAATERRLED 842
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
7-527 |
3.33e-16 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 83.45 E-value: 3.33e-16
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 7 QRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQK 86
Cdd:COG1196 235 RELEAELEELEAELEELEAELEELEAELAELEAELEELRLELEELELELEEAQAEEYELLAELARLEQDIARLEERRREL 314
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 87 LAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERE 166
Cdd:COG1196 315 EERLEELEEELAELEEELEELEEELEELEEELEEAEEELEEAEAELAEAEEALLEAEAELAEAEEELEELAEELLEALRA 394
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 167 TLQSKDFKLEVEK--------LSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESR 238
Cdd:COG1196 395 AAELAAQLEELEEaeeallerLERLEEELEELEEALAELEEEEEEEEEALEEAAEEEAELEEEEEALLELLAELLEEAAL 474
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 239 LEKTEFTLKEDLTKLKTLTVMFVDERKTMSE-----KLKKTEDKLQAASSQLQVEQnKVTTVTEKLIEETKRALkSKTDV 313
Cdd:COG1196 475 LEAALAELLEELAEAAARLLLLLEAEADYEGflegvKAALLLAGLRGLAGAVAVLI-GVEAAYEAALEAALAAA-LQNIV 552
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 314 EEKMYSVTKERDDLK------------NKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQE 381
Cdd:COG1196 553 VEDDEVAAAAIEYLKaakagratflplDKIRARAALAAALARGAIGAAVDLVASDLREADARYYVLGDTLLGRTLVAARL 632
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 382 NNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWL 461
Cdd:COG1196 633 EAALRRAVTLAGRLREVTLEGEGGSAGGSLTGGSRRELLAALLEAEAELEELAERLAEEELELEEALLAEEEEERELAEA 712
|
490 500 510 520 530 540
....*....|....*....|....*....|....*....|....*....|....*....|....*..
gi 544346182 462 FKRLQEEEAKSGHLSREVDALKEKIHEYMATEDLICHLQGDHSVLQK-KLNQQENRNRDLGREIENL 527
Cdd:COG1196 713 EEERLEEELEEEALEEQLEAEREELLEELLEEEELLEEEALEELPEPpDLEELERELERLEREIEAL 779
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
81-532 |
7.90e-16 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 82.42 E-value: 7.90e-16
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 81 RQLQQKLAALSRQIDELEET-------NRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELR---KRVLDMEGKDEELI 150
Cdd:PRK03918 217 PELREELEKLEKEVKELEELkeeieelEKELESLEGSKRKLEEKIRELEERIEELKKEIEELEekvKELKELKEKAEEYI 296
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 151 KMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLE------KERMTTKQLSQELES 224
Cdd:PRK03918 297 KLSEFYEEYLDELREIEKRLSRLEEEINGIEERIKELEEKEERLEELKKKLKELEKRLEeleerhELYEEAKAKKEELER 376
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 225 LKVRIK--ELEAIESRLEKTEFTLKEDLTKLKTLTVMF------VDERKTMSEKLKKTEDKLQAASSQLQVEQNKvtTVT 296
Cdd:PRK03918 377 LKKRLTglTPEKLEKELEELEKAKEEIEEEISKITARIgelkkeIKELKKAIEELKKAKGKCPVCGRELTEEHRK--ELL 454
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 297 EKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEE--KGNDLLSRVNMLKNRLQS--LEAIEKDF-----LKNKL 367
Cdd:PRK03918 455 EEYTAELKRIEKELKEIEEKERKLRKELRELEKVLKKESEliKLKELAEQLKELEEKLKKynLEELEKKAeeyekLKEKL 534
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 368 NQDSGKSTTaLHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDE-----YETLERR------YANERDKAQFLSKE 436
Cdd:PRK03918 535 IKLKGEIKS-LKKELEKLEELKKKLAELEKKLDELEEELAELLKELEElgfesVEELEERlkelepFYNEYLELKDAEKE 613
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 437 LEHVKMELAkyKLAEKTETSheqwlFKRLQEEEAKSGHLSREVDALKEKI--HEYMATEDLICHLQGDHSVLQKKLNQQE 514
Cdd:PRK03918 614 LEREEKELK--KLEEELDKA-----FEELAETEKRLEELRKELEELEKKYseEEYEELREEYLELSRELAGLRAELEELE 686
|
490
....*....|....*...
gi 544346182 515 NRNRDLGREIENLTKELE 532
Cdd:PRK03918 687 KRREEIKKTLEKLKEELE 704
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
66-532 |
7.00e-15 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 79.34 E-value: 7.00e-15
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 66 QDTIMAKLTNEDSQNRQLQQKLaalsrQIDELEETNRSLRKAEEELQDIKEKISKgeygnagIMAEVEELRKRVLDMEgk 145
Cdd:PRK03918 134 QGEIDAILESDESREKVVRQIL-----GLDDYENAYKNLGEVIKEIKRRIERLEK-------FIKRTENIEELIKEKE-- 199
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 146 dEELIKMEEQCRDLNKRLEretlqskDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESL 225
Cdd:PRK03918 200 -KELEEVLREINEISSELP-------ELREELEKLEKEVKELEELKEEIEELEKELESLEGSKRKLEEKIRELEERIEEL 271
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 226 KVRIKELEAIESRLEKTEftlkedltklktltvmfvdERKTMSEKLKKTEDKLQAASSQLQVEQNKVttvtEKLIEETKR 305
Cdd:PRK03918 272 KKEIEELEEKVKELKELK-------------------EKAEEYIKLSEFYEEYLDELREIEKRLSRL----EEEINGIEE 328
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 306 ALKSKTDVEEKMYSVTKERDDLKNKLkAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKI 385
Cdd:PRK03918 329 RIKELEEKEERLEELKKKLKELEKRL-EELEERHELYEEAKAKKEELERLKKRLTGLTPEKLEKELEELEKAKEEIEEEI 407
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 386 KELSQEVERLKLKLKDMKAIEDDLMK------------TEDEYETLERRY-------ANERDKAQFLSKELEHVKMELAK 446
Cdd:PRK03918 408 SKITARIGELKKEIKELKKAIEELKKakgkcpvcgrelTEEHRKELLEEYtaelkriEKELKEIEEKERKLRKELRELEK 487
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 447 yKLAEKTETSHEQWLFKRLQEEEAKSGHLSREvdALKEKIHEYMATEDLICHLQGDHSVLQ---KKLNQQENRNRDLGRE 523
Cdd:PRK03918 488 -VLKKESELIKLKELAEQLKELEEKLKKYNLE--ELEKKAEEYEKLKEKLIKLKGEIKSLKkelEKLEELKKKLAELEKK 564
|
....*....
gi 544346182 524 IENLTKELE 532
Cdd:PRK03918 565 LDELEEELA 573
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
36-451 |
2.02e-14 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 77.79 E-value: 2.02e-14
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 36 LAEARVQEEEQKATRLEKELQTQTTKFHQDQDTI-----MAKLTNEDSQNRQ-LQQKLAALSRQIDELEETNRSLRKaee 109
Cdd:TIGR02168 622 LGGVLVVDDLDNALELAKKLRPGYRIVTLDGDLVrpggvITGGSAKTNSSILeRRREIEELEEKIEELEEKIAELEK--- 698
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 110 ELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEgkdEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRimaLEK 189
Cdd:TIGR02168 699 ALAELRKELEELEEELEQLRKELEELSRQISALR---KDLARLEAEVEQLEERIAQLSKELTELEAEIEELEER---LEE 772
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 190 LEDAFNKSKQEcyslkcnLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSE 269
Cdd:TIGR02168 773 AEEELAEAEAE-------IEELEAQIEQLKEELKALREALDELRAELTLLNEEAANLRERLESLERRIAATERRLEDLEE 845
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 270 KLKKTEDKLQAASSqlqvEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLL------- 342
Cdd:TIGR02168 846 QIEELSEDIESLAA----EIEELEELIEELESELEALLNERASLEEALALLRSELEELSEELRELESKRSELRreleelr 921
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 343 SRVNMLKNRLQSLEAiEKDFLKNKLNQDsGKSTTALHQEN-----NKIKELSQEVERLKLKLKDMKAIEddlMKTEDEYE 417
Cdd:TIGR02168 922 EKLAQLELRLEGLEV-RIDNLQERLSEE-YSLTLEEAEALenkieDDEEEARRRLKRLENKIKELGPVN---LAAIEEYE 996
|
410 420 430
....*....|....*....|....*....|....
gi 544346182 418 TLERRYanerdkaQFLSKELEHVkmELAKYKLAE 451
Cdd:TIGR02168 997 ELKERY-------DFLTAQKEDL--TEAKETLEE 1021
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
43-447 |
1.82e-13 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 74.72 E-value: 1.82e-13
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 43 EEEQKATRLEKELQtqttKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDEL----EETNRSLRKAEEELQDIKEKI 118
Cdd:TIGR02169 671 SEPAELQRLRERLE----GLKRELSSLQSELRRIENRLDELSQELSDASRKIGEIekeiEQLEQEEEKLKERLEELEEDL 746
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 119 SKGEYGNAGIMAEVEELRKRVLDMEgkdEELIKMEEQCRDLNKRLERETLQSKDFKL-EVEKLSKRIMA-LEKLEDAFNK 196
Cdd:TIGR02169 747 SSLEQEIENVKSELKELEARIEELE---EDLHKLEEALNDLEARLSHSRIPEIQAELsKLEEEVSRIEArLREIEQKLNR 823
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 197 SKQEcyslKCNLEKERMTTKQLSQELESLKVRI-KELEAIESRLEKTEFTLKEDLTKLKTLtvmfvDERKtmsEKLKKTE 275
Cdd:TIGR02169 824 LTLE----KEYLEKEIQELQEQRIDLKEQIKSIeKEIENLNGKKEELEEELEELEAALRDL-----ESRL---GDLKKER 891
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 276 DKLQAASSQLQVEQNKvttvteklieetkralksktdveekmysvtkerddlknkLKAEEEKGNDLLSRvnmLKNRLQSL 355
Cdd:TIGR02169 892 DELEAQLRELERKIEE---------------------------------------LEAQIEKKRKRLSE---LKAKLEAL 929
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 356 EAIEKDFLKNKlnqDSGKSTTALHQENNKIKELSQEVERlklklkDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSK 435
Cdd:TIGR02169 930 EEELSEIEDPK---GEDEEIPEEELSLEDVQAELQRVEE------EIRALEPVNMLAIQEYEEVLKRLDELKEKRAKLEE 1000
|
410
....*....|..
gi 544346182 436 ELEHVKMELAKY 447
Cdd:TIGR02169 1001 ERKAILERIEEY 1012
|
|
| PTZ00121 |
PTZ00121 |
MAEBL; Provisional |
38-559 |
2.09e-13 |
|
MAEBL; Provisional
Pssm-ID: 173412 [Multi-domain] Cd Length: 2084 Bit Score: 74.79 E-value: 2.09e-13
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 38 EARVQEEEQKATRLEKelqTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRslRKAEEeLQDIKEK 117
Cdd:PTZ00121 1216 EARKAEDAKKAEAVKK---AEEAKKDAEEAKKAEEERNNEEIRKFEEARMAHFARRQAAIKAEEA--RKADE-LKKAEEK 1289
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 118 ISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEE---QCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAF 194
Cdd:PTZ00121 1290 KKADEAKKAEEKKKADEAKKKAEEAKKADEAKKKAEEakkKADAAKKKAEEAKKAAEAAKAEAEAAADEAEAAEEKAEAA 1369
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 195 NKSKQECYSLKCNLEK---ERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLK--EDLTKLKTLTVMfVDERKTMSE 269
Cdd:PTZ00121 1370 EKKKEEAKKKADAAKKkaeEKKKADEAKKKAEEDKKKADELKKAAAAKKKADEAKKkaEEKKKADEAKKK-AEEAKKADE 1448
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 270 KLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLK 349
Cdd:PTZ00121 1449 AKKKAEEAKKAEEAKKKAEEAKKADEAKKKAEEAKKADEAKKKAEEAKKKADEAKKAAEAKKKADEAKKAEEAKKADEAK 1528
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 350 NRLQSLEAIEKDFLKNKLNQDSGKSTTALH--QENNKIKELSQEVERLKLKLK---DMKAIE----DDLMKTEDEYETLE 420
Cdd:PTZ00121 1529 KAEEAKKADEAKKAEEKKKADELKKAEELKkaEEKKKAEEAKKAEEDKNMALRkaeEAKKAEeariEEVMKLYEEEKKMK 1608
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 421 ----RRYANERDKAQFLSKElEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKEKIHEYMATEDli 496
Cdd:PTZ00121 1609 aeeaKKAEEAKIKAEELKKA-EEEKKKVEQLKKKEAEEKKKAEELKKAEEENKIKAAEEAKKAEEDKKKAEEAKKAEE-- 1685
|
490 500 510 520 530 540
....*....|....*....|....*....|....*....|....*....|....*....|...
gi 544346182 497 chlqgdhsvLQKKLNQQENRNRDLGREIENLTKELERYRHFSKSLRPSLNGRRISDPQVFSKE 559
Cdd:PTZ00121 1686 ---------DEKKAAEALKKEAEEAKKAEELKKKEAEEKKKAEELKKAEEENKIKAEEAKKEA 1739
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
4-327 |
6.61e-13 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 72.78 E-value: 6.61e-13
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKE---LQTQTTKFHQDQDTIMAKLTNEDSQN 80
Cdd:TIGR02168 705 KELEELEEELEQLRKELEELSRQISALRKDLARLEAEVEQLEERIAQLSKElteLEAEIEELEERLEEAEEELAEAEAEI 784
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 81 RQLQQKLAALSRQIDELEETnrsLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEgkdEELIKMEEQCRDLN 160
Cdd:TIGR02168 785 EELEAQIEQLKEELKALREA---LDELRAELTLLNEEAANLRERLESLERRIAATERRLEDLE---EQIEELSEDIESLA 858
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 161 KRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELEslkvriKELEAIESRLE 240
Cdd:TIGR02168 859 AEIEELEELIEELESELEALLNERASLEEALALLRSELEELSEELRELESKRSELRRELEELR------EKLAQLELRLE 932
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 241 KTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKralksktDVEEKMYSV 320
Cdd:TIGR02168 933 GLEVRIDNLQERLSEEYSLTLEEAEALENKIEDDEEEARRRLKRLENKIKELGPVNLAAIEEYE-------ELKERYDFL 1005
|
....*..
gi 544346182 321 TKERDDL 327
Cdd:TIGR02168 1006 TAQKEDL 1012
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
1-337 |
3.77e-12 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 70.48 E-value: 3.77e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 1 MVVDEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQ---DQDTIMAKLTNED 77
Cdd:TIGR02169 678 RLRERLEGLKRELSSLQSELRRIENRLDELSQELSDASRKIGEIEKEIEQLEQEEEKLKERLEEleeDLSSLEQEIENVK 757
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 78 SQNRQLQQKLAALSRQIDELEETNRSL--RKAEEELQDIKEKISKgeygnagIMAEVEELRKRVLDMEGKDEELIKMEEQ 155
Cdd:TIGR02169 758 SELKELEARIEELEEDLHKLEEALNDLeaRLSHSRIPEIQAELSK-------LEEEVSRIEARLREIEQKLNRLTLEKEY 830
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 156 CRDLNKRLERE----TLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERmttKQLSQELESLKVRIKE 231
Cdd:TIGR02169 831 LEKEIQELQEQridlKEQIKSIEKEIENLNGKKEELEEELEELEAALRDLESRLGDLKKER---DELEAQLRELERKIEE 907
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 232 LEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKT--EDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKS 309
Cdd:TIGR02169 908 LEAQIEKKRKRLSELKAKLEALEEELSEIEDPKGEDEEIPEEElsLEDVQAELQRVEEEIRALEPVNMLAIQEYEEVLKR 987
|
330 340
....*....|....*....|....*...
gi 544346182 310 KTDVEEKMYSVTKERDDLKNKLKAEEEK 337
Cdd:TIGR02169 988 LDELKEKRAKLEEERKAILERIEEYEKK 1015
|
|
| PTZ00121 |
PTZ00121 |
MAEBL; Provisional |
17-488 |
4.49e-12 |
|
MAEBL; Provisional
Pssm-ID: 173412 [Multi-domain] Cd Length: 2084 Bit Score: 70.56 E-value: 4.49e-12
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 17 RQKIQELTTNAKETHTKlalAEARVQEEEQKATRLEK--------ELQTQTTKFHQDQdtiMAKLTNEDSQNRQLQQKLA 88
Cdd:PTZ00121 1328 KKKADAAKKKAEEAKKA---AEAAKAEAEAAADEAEAaeekaeaaEKKKEEAKKKADA---AKKKAEEKKKADEAKKKAE 1401
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 89 ALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCR--DLNKRLERE 166
Cdd:PTZ00121 1402 EDKKKADELKKAAAAKKKADEAKKKAEEKKKADEAKKKAEEAKKADEAKKKAEEAKKAEEAKKKAEEAKkaDEAKKKAEE 1481
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 167 TLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQecysLKCNLEKERMTTKQLSQEleslKVRIKELEAIESRLEKTEFTL 246
Cdd:PTZ00121 1482 AKKADEAKKKAEEAKKKADEAKKAAEAKKKADE----AKKAEEAKKADEAKKAEE----AKKADEAKKAEEKKKADELKK 1553
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 247 KEDLTKlktltvmfVDERKTMSEKLKKTEDK---LQAASSQLQVEQNKVTTVTEKLIEETK-RALKSKTDVEEKMYSvtk 322
Cdd:PTZ00121 1554 AEELKK--------AEEKKKAEEAKKAEEDKnmaLRKAEEAKKAEEARIEEVMKLYEEEKKmKAEEAKKAEEAKIKA--- 1622
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 323 erddlkNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDsgksttalhQENNKIKELSQEVERLKLKLKDM 402
Cdd:PTZ00121 1623 ------EELKKAEEEKKKVEQLKKKEAEEKKKAEELKKAEEENKIKAA---------EEAKKAEEDKKKAEEAKKAEEDE 1687
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 403 KAIEDDLMKTEDE---YETLERRYANERDKAQFLSKELE--HVKMELAKYKLAEKTETSHEqwlFKRLQEEEAKSGHLSR 477
Cdd:PTZ00121 1688 KKAAEALKKEAEEakkAEELKKKEAEEKKKAEELKKAEEenKIKAEEAKKEAEEDKKKAEE---AKKDEEEKKKIAHLKK 1764
|
490
....*....|.
gi 544346182 478 EVDALKEKIHE 488
Cdd:PTZ00121 1765 EEEKKAEEIRK 1775
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
16-348 |
1.16e-11 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 68.93 E-value: 1.16e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 16 QRQKIQELTtnakethTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTnedsQNRQLQQKLAALSRQID 95
Cdd:TIGR02168 675 RRREIEELE-------EKIEELEEKIAELEKALAELRKELEELEEELEQLRKELEELSR----QISALRKDLARLEAEVE 743
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 96 ELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRV-LDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFK 174
Cdd:TIGR02168 744 QLEERIAQLSKELTELEAEIEELEERLEEAEEELAEAEAEIEELeAQIEQLKEELKALREALDELRAELTLLNEEAANLR 823
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 175 LEVEKLSKRIMALEK----LEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDL 250
Cdd:TIGR02168 824 ERLESLERRIAATERrledLEEQIEELSEDIESLAAEIEELEELIEELESELEALLNERASLEEALALLRSELEELSEEL 903
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 251 TKLKtltvmfvDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKsktDVEEKMYSVTKERDDLKNK 330
Cdd:TIGR02168 904 RELE-------SKRSELRRELEELREKLAQLELRLEGLEVRIDNLQERLSEEYSLTLE---EAEALENKIEDDEEEARRR 973
|
330
....*....|....*...
gi 544346182 331 LKAEEEKGNDlLSRVNML 348
Cdd:TIGR02168 974 LKRLENKIKE-LGPVNLA 990
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
94-468 |
1.18e-11 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 68.94 E-value: 1.18e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 94 IDEL---EETNRSLRKAEEELQDIKEKISKgeygnagIMAEVEELRKRVLDMEGKDEELIKMeeqcRDLNKRLEretlqs 170
Cdd:TIGR02169 159 IDEIagvAEFDRKKEKALEELEEVEENIER-------LDLIIDEKRQQLERLRREREKAERY----QALLKEKR------ 221
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 171 kdfKLEVEKLSKRIMALEKledafnkskqecyslkcnlEKERmTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDL 250
Cdd:TIGR02169 222 ---EYEGYELLKEKEALER-------------------QKEA-IERQLASLEEELEKLTEEISELEKRLEEIEQLLEELN 278
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 251 TKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNK 330
Cdd:TIGR02169 279 KKIKDLGEEEQLRVKEKIGELEAEIASLERSIAEKERELEDAEERLAKLEAEIDKLLAEIEELEREIEEERKRRDKLTEE 358
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 331 LKAEEEKGNDLLSRVNMLKNRLQSL------EAIEKDFLKNKLN----------QDSGKSTTALHQENNKIKELSQEV-- 392
Cdd:TIGR02169 359 YAELKEELEDLRAELEEVDKEFAETrdelkdYREKLEKLKREINelkreldrlqEELQRLSEELADLNAAIAGIEAKIne 438
|
330 340 350 360 370 380 390
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*...
gi 544346182 393 --ERLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKyKLAEKTETSHEQWLFKRLQEE 468
Cdd:TIGR02169 439 leEEKEDKALEIKKQEWKLEQLAADLSKYEQELYDLKEEYDRVEKELSKLQRELAE-AEAQARASEERVRGGRAVEEV 515
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
6-358 |
1.27e-11 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 68.53 E-value: 1.27e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 6 QQRLTAQLTLQRQKIQELTTNAKETHTKLA-----LAEARVQEEEQKATR--LEKELQTQTTKFhqdqDTIMAKLTNEDS 78
Cdd:PRK02224 337 AQAHNEEAESLREDADDLEERAEELREEAAeleseLEEAREAVEDRREEIeeLEEEIEELRERF----GDAPVDLGNAED 412
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 79 QNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYG----NAGIMAEVEELRKRVLDMEgkdEELIKMEE 154
Cdd:PRK02224 413 FLEELREERDELREREAELEATLRTARERVEEAEALLEAGKCPECGqpveGSPHVETIEEDRERVEELE---AELEDLEE 489
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 155 QCRDLNKRLER-----------ETLQSK---------DFKLEVEKLSKRIMAL----EKLEDAFNKSKQECYSLKCNLEK 210
Cdd:PRK02224 490 EVEEVEERLERaedlveaedriERLEERredleeliaERRETIEEKRERAEELreraAELEAEAEEKREAAAEAEEEAEE 569
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 211 ERMTTKQLSQELESLKVRIKELEAIESRLEKTEfTLKEDLTKLKtltvmfvDERKTMSEKLKKTEDKLQAAS---SQL-- 285
Cdd:PRK02224 570 AREEVAELNSKLAELKERIESLERIRTLLAAIA-DAEDEIERLR-------EKREALAELNDERRERLAEKRerkRELea 641
|
330 340 350 360 370 380 390
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 286 QVEQNKVttvtEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNK---LKAEEEKGNDLLSRVNMLKNRLQSLEAI 358
Cdd:PRK02224 642 EFDEARI----EEAREDKERAEEYLEQVEEKLDELREERDDLQAEigaVENELEELEELRERREALENRVEALEAL 713
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
3-448 |
1.38e-11 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 68.26 E-value: 1.38e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 3 VDEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTkfHQDQDTIMAKLTNEDSQNRQ 82
Cdd:COG4717 73 LKELEEELKEAEEKEEEYAELQEELEELEEELEELEAELEELREELEKLEKLLQLLPL--YQELEALEAELAELPERLEE 150
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 83 LQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAE-VEELRKRVLDMEgkdEELIKMEEQCRDLNK 161
Cdd:COG4717 151 LEERLEELRELEEELEELEAELAELQEELEELLEQLSLATEEELQDLAEeLEELQQRLAELE---EELEEAQEELEELEE 227
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 162 RLERETLQSKDFKLEvEKLSKR---------IMALEKLEDAFNKSKQEC-----------YSLKCNLEKERMTTKQLSQE 221
Cdd:COG4717 228 ELEQLENELEAAALE-ERLKEArlllliaaaLLALLGLGGSLLSLILTIagvlflvlgllALLFLLLAREKASLGKEAEE 306
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 222 LESLKvRIKELEAIESRLEKTEFTLKEDLTKLktltvmFVDERKTMSEKLKKTEDKLQAASSQLQVEQnkvttvteklIE 301
Cdd:COG4717 307 LQALP-ALEELEEEELEELLAALGLPPDLSPE------ELLELLDRIEELQELLREAEELEEELQLEE----------LE 369
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 302 ETKRALKSKTDVEekmysvtkERDDLKNKLKAEEEKgNDLLSRVNMLKNRLQSL----EAIEKDFLKNKLNQDSGKSTTA 377
Cdd:COG4717 370 QEIAALLAEAGVE--------DEEELRAALEQAEEY-QELKEELEELEEQLEELlgelEELLEALDEEELEEELEELEEE 440
|
410 420 430 440 450 460 470
....*....|....*....|....*....|....*....|....*....|....*....|....*....|.
gi 544346182 378 LHQENNKIKELSQEVERLKLKLKDMKAiEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYK 448
Cdd:COG4717 441 LEELEEELEELREELAELEAELEQLEE-DGELAELLQELEELKAELRELAEEWAALKLALELLEEAREEYR 510
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
3-404 |
1.61e-11 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 68.17 E-value: 1.61e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 3 VDEQQRLTAQLTLQRQKIQELTTNAKETHTKlalaEARVQEEEQKATRLEKELQtQTTKFHQDQDTIMAKLTNEdsqnRQ 82
Cdd:PRK03918 306 LDELREIEKRLSRLEEEINGIEERIKELEEK----EERLEELKKKLKELEKRLE-ELEERHELYEEAKAKKEEL----ER 376
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 83 LQQKLAALS--RQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRK---------RVLDMEGKDE---- 147
Cdd:PRK03918 377 LKKRLTGLTpeKLEKELEELEKAKEEIEEEISKITARIGELKKEIKELKKAIEELKKakgkcpvcgRELTEEHRKEllee 456
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 148 ----------ELIKMEEQCRDLNKRLER---------------------ETLQSKDFKLEVEKLSKRIMALEKLEDAFNK 196
Cdd:PRK03918 457 ytaelkriekELKEIEEKERKLRKELRElekvlkkeseliklkelaeqlKELEEKLKKYNLEELEKKAEEYEKLKEKLIK 536
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 197 SKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEF-TLKEDLTKLKTLTVMFvdeRKTMSekLKKTE 275
Cdd:PRK03918 537 LKGEIKSLKKELEKLEELKKKLAELEKKLDELEEELAELLKELEELGFeSVEELEERLKELEPFY---NEYLE--LKDAE 611
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 276 DKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKER-DDLKNKLKAEEEKGNDLLSRVNMLKNRLQS 354
Cdd:PRK03918 612 KELEREEKELKKLEEELDKAFEELAETEKRLEELRKELEELEKKYSEEEyEELREEYLELSRELAGLRAELEELEKRREE 691
|
410 420 430 440 450
....*....|....*....|....*....|....*....|....*....|
gi 544346182 355 LEAIEKDFLKNKLNQDSGKsttalhQENNKIKELSQEVERLKLKLKDMKA 404
Cdd:PRK03918 692 IKKTLEKLKEELEEREKAK------KELEKLEKALERVEELREKVKKYKA 735
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
81-438 |
2.75e-11 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 67.77 E-value: 2.75e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 81 RQLQQKLAALSRQIDELEETNRSLRKAE------EELQDIKEKISKGEYGNAGimAEVEELRKRvldMEGKDEELIKMEE 154
Cdd:TIGR02168 179 RKLERTRENLDRLEDILNELERQLKSLErqaekaERYKELKAELRELELALLV--LRLEELREE---LEELQEELKEAEE 253
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 155 QCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKledafnkskqECYSLKCNLEKERMTTKQLSQELESLKVRIKELEA 234
Cdd:TIGR02168 254 ELEELTAELQELEEKLEELRLEVSELEEEIEELQK----------ELYALANEISRLEQQKQILRERLANLERQLEELEA 323
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 235 IESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEetkralksktdVE 314
Cdd:TIGR02168 324 QLEELESKLDELAEELAELEEKLEELKEELESLEAELEELEAELEELESRLEELEEQLETLRSKVAQ-----------LE 392
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 315 EKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVER 394
Cdd:TIGR02168 393 LQIASLNNEIERLEARLERLEDRRERLQQEIEELLKKLEEAELKELQAELEELEEELEELQEELERLEEALEELREELEE 472
|
330 340 350 360
....*....|....*....|....*....|....*....|....
gi 544346182 395 LKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELE 438
Cdd:TIGR02168 473 AEQALDAAERELAQLQARLDSLERLQENLEGFSEGVKALLKNQS 516
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
12-547 |
4.88e-11 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 67.02 E-value: 4.88e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 12 QLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFH---QDQDTIMAKLTNE-DSQNRQLQQKL 87
Cdd:TIGR02169 217 LKEKREYEGYELLKEKEALERQKEAIERQLASLEEELEKLTEEISELEKRLEeieQLLEELNKKIKDLgEEEQLRVKEKI 296
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 88 AALSRQI----DELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEgkdEELIKMEEQCRDLNKRL 163
Cdd:TIGR02169 297 GELEAEIasleRSIAEKERELEDAEERLAKLEAEIDKLLAEIEELEREIEEERKRRDKLT---EEYAELKEELEDLRAEL 373
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 164 EREtlqSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEaieSRLEKTE 243
Cdd:TIGR02169 374 EEV---DKEFAETRDELKDYREKLEKLKREINELKRELDRLQEELQRLSEELADLNAAIAGIEAKINELE---EEKEDKA 447
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 244 FTLKEDLTKLKTLTVMFVDERKTM---SEKLKKTEDKLQAASSQL-------------QVEQNKVTTVTEKLIEETKRAL 307
Cdd:TIGR02169 448 LEIKKQEWKLEQLAADLSKYEQELydlKEEYDRVEKELSKLQRELaeaeaqaraseerVRGGRAVEEVLKASIQGVHGTV 527
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 308 KSKTDVEEK------------MYSVTKERDD--------LK------------NKLKAEE-------------------- 335
Cdd:TIGR02169 528 AQLGSVGERyataievaagnrLNNVVVEDDAvakeaielLKrrkagratflplNKMRDERrdlsilsedgvigfavdlve 607
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 336 ----------------------EKGNDLLSRVNM------------------------------LKNRLQSLEAiEKDFL 363
Cdd:TIGR02169 608 fdpkyepafkyvfgdtlvvediEAARRLMGKYRMvtlegelfeksgamtggsraprggilfsrsEPAELQRLRE-RLEGL 686
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 364 KNKLNqdsgksttALHQENNKIK----ELSQEVERLKLKLKDmkaIEDDLMKTEDEYETLERRYANERDKAQFLSKELEH 439
Cdd:TIGR02169 687 KRELS--------SLQSELRRIEnrldELSQELSDASRKIGE---IEKEIEQLEQEEEKLKERLEELEEDLSSLEQEIEN 755
|
570 580 590 600 610 620 630 640
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 440 VKMELAKY--KLAEKTETSHEqwLFKRLQEEEAK-SGHLSREVDALKEKIHEYMA--------TEDLICHLQGDHSVLQK 508
Cdd:TIGR02169 756 VKSELKELeaRIEELEEDLHK--LEEALNDLEARlSHSRIPEIQAELSKLEEEVSriearlreIEQKLNRLTLEKEYLEK 833
|
650 660 670 680 690
....*....|....*....|....*....|....*....|....*....|...
gi 544346182 509 KLNQQENRNRDL-------GREIENLTK-------ELERYRHFSKSLRPSLNG 547
Cdd:TIGR02169 834 EIQELQEQRIDLkeqiksiEKEIENLNGkkeeleeELEELEAALRDLESRLGD 886
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
77-369 |
7.81e-11 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 66.23 E-value: 7.81e-11
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 77 DSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQC 156
Cdd:TIGR02168 235 EELREELEELQEELKEAEEELEELTAELQELEEKLEELRLEVSELEEEIEELQKELYALANEISRLEQQKQILRERLANL 314
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 157 RDLNKRLERETLQSKDFKLEVEKlskrimALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIE 236
Cdd:TIGR02168 315 ERQLEELEAQLEELESKLDELAE------ELAELEEKLEELKEELESLEAELEELEAELEELESRLEELEEQLETLRSKV 388
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 237 SRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAAssqlqvEQNKVTTVTEKLIEETKRALKSKTDVEEK 316
Cdd:TIGR02168 389 AQLELQIASLNNEIERLEARLERLEDRRERLQQEIEELLKKLEEA------ELKELQAELEELEEELEELQEELERLEEA 462
|
250 260 270 280 290
....*....|....*....|....*....|....*....|....*....|...
gi 544346182 317 MYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQ 369
Cdd:TIGR02168 463 LEELREELEEAEQALDAAERELAQLQARLDSLERLQENLEGFSEGVKALLKNQ 515
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
9-533 |
1.37e-10 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 65.47 E-value: 1.37e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 9 LTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQ-------TTKFHQDQDTIM---AKLTNEDS 78
Cdd:TIGR02169 299 LEAEIASLERSIAEKERELEDAEERLAKLEAEIDKLLAEIEELEREIEEErkrrdklTEEYAELKEELEdlrAELEEVDK 378
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 79 QNR-------QLQQKLAALSRQIDELEETNRSL----RKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDE 147
Cdd:TIGR02169 379 EFAetrdelkDYREKLEKLKREINELKRELDRLqeelQRLSEELADLNAAIAGIEAKINELEEEKEDKALEIKKQEWKLE 458
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 148 ELIKMEEqcrDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELesLKV 227
Cdd:TIGR02169 459 QLAADLS---KYEQELYDLKEEYDRVEKELSKLQRELAEAEAQARASEERVRGGRAVEEVLKASIQGVHGTVAQL--GSV 533
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 228 RIKELEAIES----RLE----KTEFTLKEDLTKLKT----------LTVMFVDERktMSEKLKKT------------EDK 277
Cdd:TIGR02169 534 GERYATAIEVaagnRLNnvvvEDDAVAKEAIELLKRrkagratflpLNKMRDERR--DLSILSEDgvigfavdlvefDPK 611
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 278 LQAASSQ-----LQVE----------QNKVTTVTEKLIEET----------KRALKSKTDVEEKMYSVTKERDDLKNKLK 332
Cdd:TIGR02169 612 YEPAFKYvfgdtLVVEdieaarrlmgKYRMVTLEGELFEKSgamtggsrapRGGILFSRSEPAELQRLRERLEGLKRELS 691
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 333 AEEEKGNDLLSRVNMLKNRL----QSLEAIEKDflKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKA---- 404
Cdd:TIGR02169 692 SLQSELRRIENRLDELSQELsdasRKIGEIEKE--IEQLEQEEEKLKERLEELEEDLSSLEQEIENVKSELKELEAriee 769
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 405 IEDDLMKTEDEYETLERRYANERdkAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKE 484
Cdd:TIGR02169 770 LEEDLHKLEEALNDLEARLSHSR--IPEIQAELSKLEEEVSRIEARLREIEQKLNRLTLEKEYLEKEIQELQEQRIDLKE 847
|
570 580 590 600
....*....|....*....|....*....|....*....|....*....
gi 544346182 485 KIHEYMATEDLichLQGDHSVLQKKLNQQENRNRDLGREIENLTKELER 533
Cdd:TIGR02169 848 QIKSIEKEIEN---LNGKKEELEEELEELEAALRDLESRLGDLKKERDE 893
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
38-454 |
1.60e-10 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 65.06 E-value: 1.60e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 38 EARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEK 117
Cdd:PRK02224 187 GSLDQLKAQIEEKEEKDLHERLNGLESELAELDEEIERYEEQREQARETRDEADEVLEEHEERREELETLEAEIEDLRET 266
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 118 ISKGEYGNAGIMAEVEELRKRVLDMEGKD---------------------EELIKMEEQCRDlnkRLERETLQSKDFKLE 176
Cdd:PRK02224 267 IAETEREREELAEEVRDLRERLEELEEERddllaeaglddadaeavearrEELEDRDEELRD---RLEECRVAAQAHNEE 343
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 177 VEKLSKRIMALE----KLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIK----ELEAIESRLEKTEFTLKE 248
Cdd:PRK02224 344 AESLREDADDLEeraeELREEAAELESELEEAREAVEDRREEIEELEEEIEELRERFGdapvDLGNAEDFLEELREERDE 423
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 249 DLTKLKTLTVmfvdERKTMSEKLKKTEDKLQAAS--------------SQLQVEQNKVTTVTEKL--IEETKRALKSKTD 312
Cdd:PRK02224 424 LREREAELEA----TLRTARERVEEAEALLEAGKcpecgqpvegsphvETIEEDRERVEELEAELedLEEEVEEVEERLE 499
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 313 VEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAiEKDFLKNKLnQDSGKSTTALHQENNK----IKEL 388
Cdd:PRK02224 500 RAEDLVEAEDRIERLEERREDLEELIAERRETIEEKRERAEELRE-RAAELEAEA-EEKREAAAEAEEEAEEareeVAEL 577
|
410 420 430 440 450 460
....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 389 SQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRyaNERDKAQflsKELEhvkmELAKYKLAEKTE 454
Cdd:PRK02224 578 NSKLAELKERIESLERIRTLLAAIADAEDEIERL--REKREAL---AELN----DERRERLAEKRE 634
|
|
| sbcc |
TIGR00618 |
exonuclease SbcC; All proteins in this family for which functions are known are part of an ... |
4-568 |
6.13e-10 |
|
exonuclease SbcC; All proteins in this family for which functions are known are part of an exonuclease complex with sbcD homologs. This complex is involved in the initiation of recombination to regulate the levels of palindromic sequences in DNA. This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University). [DNA metabolism, DNA replication, recombination, and repair]
Pssm-ID: 129705 [Multi-domain] Cd Length: 1042 Bit Score: 63.06 E-value: 6.13e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKEthtkLALAEARVQEEEQKATRLEKelQTQTTKFHQDQDTIMAKLTNEDSQNRQL 83
Cdd:TIGR00618 253 EEQLKKQQLLKQLRARIEELRAQEAV----LEETQERINRARKAAPLAAH--IKAVTQIEQQAQRIHTELQSKMRSRAKL 326
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 84 QQKLAALSRQIDELEETNRSLRKAEEELQDIKekiskgeygnagIMAEVEELRKRVLDMEGKDEELIKMEEQcrdlnkRL 163
Cdd:TIGR00618 327 LMKRAAHVKQQSSIEEQRRLLQTLHSQEIHIR------------DAHEVATSIREISCQQHTLTQHIHTLQQ------QK 388
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 164 ERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLK--CNLEKERMTTKQL--SQELESLKVRIKELEAIESRL 239
Cdd:TIGR00618 389 TTLTQKLQSLCKELDILQREQATIDTRTSAFRDLQGQLAHAKkqQELQQRYAELCAAaiTCTAQCEKLEKIHLQESAQSL 468
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 240 EKTEFTLK------EDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLqVEQNKVTTVTEKLIEETKRALKSKTDV 313
Cdd:TIGR00618 469 KEREQQLQtkeqihLQETRKKAVVLARLLELQEEPCPLCGSCIHPNPARQDI-DNPGPLTRRMQRGEQTYAQLETSEEDV 547
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 314 EEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIK------- 386
Cdd:TIGR00618 548 YHQLTSERKQRASLKEQMQEIQQSFSILTQCDNRSKEDIPNLQNITVRLQDLTEKLSEAEDMLACEQHALLRKlqpeqdl 627
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 387 -----ELSQEVERLKLKLKDMKAIEDDLMK----------TEDEYETLERRyANERDKAQFLSKELEHVKMELAKYKLAE 451
Cdd:TIGR00618 628 qdvrlHLQQCSQELALKLTALHALQLTLTQervrehalsiRVLPKELLASR-QLALQKMQSEKEQLTYWKEMLAQCQTLL 706
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 452 KTETSHEQWLFKRLQEEEAKSG----HLSREVDALKEKIHEYMATED-LICHLQGDHSVLQKKLNQQENRN---RDLGRE 523
Cdd:TIGR00618 707 RELETHIEEYDREFNEIENASSslgsDLAAREDALNQSLKELMHQARtVLKARTEAHFNNNEEVTAALQTGaelSHLAAE 786
|
570 580 590 600
....*....|....*....|....*....|....*....|....*
gi 544346182 524 IENLTKELERYRHFSKSLRPSLNGRRISDPQVFSKEVQTEAVDNE 568
Cdd:TIGR00618 787 IQFFNRLREEDTHLLKTLEAEIGQEIPSDEDILNLQCETLVQEEE 831
|
|
| CCDC158 |
pfam15921 |
Coiled-coil domain-containing protein 158; CCDC158 is a family of proteins found in eukaryotes. ... |
4-570 |
6.52e-10 |
|
Coiled-coil domain-containing protein 158; CCDC158 is a family of proteins found in eukaryotes. The function is not known.
Pssm-ID: 464943 [Multi-domain] Cd Length: 1112 Bit Score: 63.21 E-value: 6.52e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTA-------QLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNE 76
Cdd:pfam15921 377 DQLQKLLAdlhkrekELSLEKEQNKRLWDRDTGNSITIDHLRRELDDRNMEVQRLEALLKAMKSECQGQMERQMAAIQGK 456
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 77 D-------SQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVlDMEGKDEEL 149
Cdd:pfam15921 457 NeslekvsSLTAQLESTKEMLRKVVEELTAKKMTLESSERTVSDLTASLQEKERAIEATNAEITKLRSRV-DLKLQELQH 535
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 150 IKME-EQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLK-- 226
Cdd:pfam15921 536 LKNEgDHLRNVQTECEALKLQMAEKDKVIEILRQQIENMTQLVGQHGRTAGAMQVEKAQLEKEINDRRLELQEFKILKdk 615
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 227 --VRIKELEAIESRLEKTEFTL----KEDLTKLKTLTvmfvDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLi 300
Cdd:pfam15921 616 kdAKIRELEARVSDLELEKVKLvnagSERLRAVKDIK----QERDQLLNEVKTSRNELNSLSEDYEVLKRNFRNKSEEM- 690
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 301 EETKRALKSktdveeKMYSVTKERDDLKNKLKAEEEKGNDLLS--------------RVNMLKNRLQSLEAI------EK 360
Cdd:pfam15921 691 ETTTNKLKM------QLKSAQSELEQTRNTLKSMEGSDGHAMKvamgmqkqitakrgQIDALQSKIQFLEEAmtnankEK 764
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 361 DFLKNKLNQDSGKSTTALHQENNKIKEL----SQEvERLKLKLKDMKAIEDDLMKTEDEYETLERRYANE--RDKAQFL- 433
Cdd:pfam15921 765 HFLKEEKNKLSQELSTVATEKNKMAGELevlrSQE-RRLKEKVANMEVALDKASLQFAECQDIIQRQEQEsvRLKLQHTl 843
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 434 -SKELE---HVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKEKiheymATEDLICHLQGDHSVLQKK 509
Cdd:pfam15921 844 dVKELQgpgYTSNSSMKPRLLQPASFTRTHSNVPSSQSTASFLSHHSRKTNALKED-----PTRDLKQLLQELRSVINEE 918
|
570 580 590 600 610 620 630
....*....|....*....|....*....|....*....|....*....|....*....|....*....|.
gi 544346182 510 LNQQENRNRDLGRE---------IENLTKELERYRHFSKSLRPSLNGRRISDPQVFSKE-VQTEAVDNEPP 570
Cdd:pfam15921 919 PTVQLSKAEDKGRApslgalddrVRDCIIESSLRSDICHSSSNSLQTEGSKSSETCSREpVLLHAGELEDP 989
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
85-496 |
9.81e-10 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 62.09 E-value: 9.81e-10
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 85 QKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEygnagimAEVEELRKRVLDMEgKDEELIKMEEQCRDLNKRLE 164
Cdd:COG4717 71 KELKELEEELKEAEEKEEEYAELQEELEELEEELEELE-------AELEELREELEKLE-KLLQLLPLYQELEALEAELA 142
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 165 RETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNL-EKERMTTKQLSQELESLKVRIKELEAIESRLEKTE 243
Cdd:COG4717 143 ELPERLEELEERLEELRELEEELEELEAELAELQEELEELLEQLsLATEEELQDLAEELEELQQRLAELEEELEEAQEEL 222
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 244 FTLKEDLTKLKTLTvmfvdERKTMSEKLKKTEDKLQAASSQLQVE------------------------------QNKVT 293
Cdd:COG4717 223 EELEEELEQLENEL-----EAAALEERLKEARLLLLIAAALLALLglggsllsliltiagvlflvlgllallfllLAREK 297
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 294 TVTEKLIEETkRALKSKTDVEEKMYSVTKERDDLKNKLKAEE--------EKGNDLLSRVNMLKNRLQ-SLEAIEKDFLK 364
Cdd:COG4717 298 ASLGKEAEEL-QALPALEELEEEELEELLAALGLPPDLSPEEllelldriEELQELLREAEELEEELQlEELEQEIAALL 376
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 365 NKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEyETLERRYANERDKAQFLSKELEHVKMEL 444
Cdd:COG4717 377 AEAGVEDEEELRAALEQAEEYQELKEELEELEEQLEELLGELEELLEALDE-EELEEELEELEEELEELEEELEELREEL 455
|
410 420 430 440 450
....*....|....*....|....*....|....*....|....*....|...
gi 544346182 445 AKYKLA-EKTETSHEqwLFKRLQEEEaksgHLSREVDALKEKIHEYMATEDLI 496
Cdd:COG4717 456 AELEAElEQLEEDGE--LAELLQELE----ELKAELRELAEEWAALKLALELL 502
|
|
| PRK02224 |
PRK02224 |
DNA double-strand break repair Rad50 ATPase; |
5-488 |
1.31e-09 |
|
DNA double-strand break repair Rad50 ATPase;
Pssm-ID: 179385 [Multi-domain] Cd Length: 880 Bit Score: 61.98 E-value: 1.31e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 5 EQQRLTAQLTLQR------------QKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELqtqttkfhqdqDTIMAK 72
Cdd:PRK02224 233 RETRDEADEVLEEheerreeletleAEIEDLRETIAETEREREELAEEVRDLRERLEELEEER-----------DDLLAE 301
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 73 LTNEDSQNRQLQQKLAALSRQIDELEETnrslrkAEEELQDIKEKISkgeygnagimaEVEELRKRVLDMEGKDEELikm 152
Cdd:PRK02224 302 AGLDDADAEAVEARREELEDRDEELRDR------LEECRVAAQAHNE-----------EAESLREDADDLEERAEEL--- 361
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 153 EEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEK----LEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVR 228
Cdd:PRK02224 362 REEAAELESELEEAREAVEDRREEIEELEEEIEELRErfgdAPVDLGNAEDFLEELREERDELREREAELEATLRTARER 441
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 229 IKE----------------------LEAIESRLEKTEfTLKEDLTKLKTlTVMFVDERKTMSEKLKKTE-------DKLQ 279
Cdd:PRK02224 442 VEEaealleagkcpecgqpvegsphVETIEEDRERVE-ELEAELEDLEE-EVEEVEERLERAEDLVEAEdrierleERRE 519
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 280 AASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDdlknklKAEE--EKGNDLLSRVNMLKNRLQSLEA 357
Cdd:PRK02224 520 DLEELIAERRETIEEKRERAEELRERAAELEAEAEEKREAAAEAEE------EAEEarEEVAELNSKLAELKERIESLER 593
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 358 IEKDFlknklnqdsgksttalhqenNKIKELSQEVERLKLKLKDMKAIEDdlmkteDEYETLERRyaneRDKAQFLSKEL 437
Cdd:PRK02224 594 IRTLL--------------------AAIADAEDEIERLREKREALAELND------ERRERLAEK----RERKRELEAEF 643
|
490 500 510 520 530
....*....|....*....|....*....|....*....|....*....|.
gi 544346182 438 EHVKMELAKYKLaEKTETSHEQwLFKRLQEEEAKSGHLSREVDALKEKIHE 488
Cdd:PRK02224 644 DEARIEEAREDK-ERAEEYLEQ-VEEKLDELREERDDLQAEIGAVENELEE 692
|
|
| DUF3584 |
pfam12128 |
Protein of unknown function (DUF3584); This protein is found in bacteria and eukaryotes. ... |
39-537 |
1.97e-09 |
|
Protein of unknown function (DUF3584); This protein is found in bacteria and eukaryotes. Proteins in this family are typically between 943 to 1234 amino acids in length. This family contains a P-loop motif suggesting it is a nucleotide binding protein. It may be involved in replication.
Pssm-ID: 432349 [Multi-domain] Cd Length: 1191 Bit Score: 61.78 E-value: 1.97e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 39 ARVQEEEQKATRLEKELQT--------QTTKFH--QDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDEL-EETNRSLRKA 107
Cdd:pfam12128 234 AGIMKIRPEFTKLQQEFNTlesaelrlSHLHFGykSDETLIASRQEERQETSAELNQLLRTLDDQWKEKrDELNGELSAA 313
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 108 EEELQDIKEKIS-----KGEYGNAGI---MAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLE-RETLQSKDFKLEVE 178
Cdd:pfam12128 314 DAAVAKDRSELEaledqHGAFLDADIetaAADQEQLPSWQSELENLEERLKALTGKHQDVTAKYNrRRSKIKEQNNRDIA 393
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 179 KLSKRimaLEKLEDAFNKSKQECYSLKCNLEKErmttkqLSQELESLKVRIKELE-AIESRLEKTEFTLKEDLTKLKTLT 257
Cdd:pfam12128 394 GIKDK---LAKIREARDRQLAVAEDDLQALESE------LREQLEAGKLEFNEEEyRLKSRLGELKLRLNQATATPELLL 464
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 258 VM--FVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMY----------------- 318
Cdd:pfam12128 465 QLenFDERIERAREEQEAANAEVERLQSELRQARKRRDQASEALRQASRRLEERQSALDELELqlfpqagtllhflrkea 544
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 319 -----SVTK-------ERDDLKNKLKAEEEKGNDLLSRVNMlknRLQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIK 386
Cdd:pfam12128 545 pdweqSIGKvispellHRTDLDPEVWDGSVGGELNLYGVKL---DLKRIDVPEWAASEEELRERLDKAEEALQSAREKQA 621
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 387 ELSQ-------EVERLKLKLKDMKAI----EDDLMKTEDEYETLERR----YANERDKAQFLSKELEHVKMELA---KYK 448
Cdd:pfam12128 622 AAEEqlvqangELEKASREETFARTAlknaRLDLRRLFDEKQSEKDKknkaLAERKDSANERLNSLEAQLKQLDkkhQAW 701
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 449 LAEKTETSHE---QWLFKRLQEEEAKSGHLSReVDALKEKIHEYMATEDLICHLQGDHSVlqKKLNQQENRNRDLGREIE 525
Cdd:pfam12128 702 LEEQKEQKREartEKQAYWQVVEGALDAQLAL-LKAAIAARRSGAKAELKALETWYKRDL--ASLGVDPDVIAKLKREIR 778
|
570
....*....|..
gi 544346182 526 NLTKELERYRHF 537
Cdd:pfam12128 779 TLERKIERIAVR 790
|
|
| PRK05771 |
PRK05771 |
V-type ATP synthase subunit I; Validated |
174-438 |
3.90e-09 |
|
V-type ATP synthase subunit I; Validated
Pssm-ID: 235600 [Multi-domain] Cd Length: 646 Bit Score: 60.33 E-value: 3.90e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 174 KLEVEKLSKRIMALEKLEDAFNKSKQecYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKteftLKEDLTKL 253
Cdd:PRK05771 39 ELSNERLRKLRSLLTKLSEALDKLRS--YLPKLNPLREEKKKVSVKSLEELIKDVEEELEKIEKEIKE----LEEEISEL 112
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 254 KtltvmfvDERKTMSEKLKKTEdKLQAASSQLQVEQN-KVTTVTEKLIEETKralksktdveekmYSVTKERDDLKNKLK 332
Cdd:PRK05771 113 E-------NEIKELEQEIERLE-PWGNFDLDLSLLLGfKYVSVFVGTVPEDK-------------LEELKLESDVENVEY 171
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 333 AEEEKGNDLLSRVNmLKNRLQSLEAI--EKDFLKNKLNqDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLM 410
Cdd:PRK05771 172 ISTDKGYVYVVVVV-LKELSDEVEEElkKLGFERLELE-EEGTPSELIREIKEELEEIEKERESLLEELKELAKKYLEEL 249
|
250 260
....*....|....*....|....*...
gi 544346182 411 KTEDEYetlerrYANERDKAQFLSKELE 438
Cdd:PRK05771 250 LALYEY------LEIELERAEALSKFLK 271
|
|
| PRK03918 |
PRK03918 |
DNA double-strand break repair ATPase Rad50; |
291-546 |
7.29e-09 |
|
DNA double-strand break repair ATPase Rad50;
Pssm-ID: 235175 [Multi-domain] Cd Length: 880 Bit Score: 59.69 E-value: 7.29e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 291 KVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAI-----EKDFLKN 365
Cdd:PRK03918 169 EVIKEIKRRIERLEKFIKRTENIEELIKEKEKELEEVLREINEISSELPELREELEKLEKEVKELEELkeeieELEKELE 248
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 366 KLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEddlmKTEDEYETLERRYANERDKAQFLSKELEHVKMELA 445
Cdd:PRK03918 249 SLEGSKRKLEEKIRELEERIEELKKEIEELEEKVKELKELK----EKAEEYIKLSEFYEEYLDELREIEKRLSRLEEEIN 324
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 446 --KYKLAEKTETSheqwlfKRLQEEEAKSGHLSREVDALKEKIHEYMATEDLICHLQGDHSV--------LQKKLNQQEN 515
Cdd:PRK03918 325 giEERIKELEEKE------ERLEELKKKLKELEKRLEELEERHELYEEAKAKKEELERLKKRltgltpekLEKELEELEK 398
|
250 260 270
....*....|....*....|....*....|.
gi 544346182 516 RNRDLGREIENLTKELERYRHFSKSLRPSLN 546
Cdd:PRK03918 399 AKEEIEEEISKITARIGELKKEIKELKKAIE 429
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
2-535 |
7.37e-09 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 59.69 E-value: 7.37e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 2 VVDEQQRLTAQLTLQRQKIQELTTNAKETHTKLalaEARVQEEEQKATRLEKELQTQTTKFHQDQ-DTIMAKLTNEDSQN 80
Cdd:TIGR02168 380 QLETLRSKVAQLELQIASLNNEIERLEARLERL---EDRRERLQQEIEELLKKLEEAELKELQAElEELEEELEELQEEL 456
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 81 RQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDE---ELIKMEEQCR 157
Cdd:TIGR02168 457 ERLEEALEELREELEEAEQALDAAERELAQLQARLDSLERLQENLEGFSEGVKALLKNQSGLSGILGvlsELISVDEGYE 536
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 158 -----DLNKRL-----ERETLQSKDFKLEVEKLSKRIM----------ALEKLEDAFNKSKQECYSLKCNLEKermTTKQ 217
Cdd:TIGR02168 537 aaieaALGGRLqavvvENLNAAKKAIAFLKQNELGRVTflpldsikgtEIQGNDREILKNIEGFLGVAKDLVK---FDPK 613
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 218 LSQELESLKVRIKELEAIESRLEK--------TEFTLKEDL-------TKLKTLTVMFVDERKTMSEKLKKTEDKLQAAS 282
Cdd:TIGR02168 614 LRKALSYLLGGVLVVDDLDNALELakklrpgyRIVTLDGDLvrpggviTGGSAKTNSSILERRREIEELEEKIEELEEKI 693
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 283 SQLQVEQNKVTTVTEKLIEETKRALKSKTDVEekmysvtKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEkdf 362
Cdd:TIGR02168 694 AELEKALAELRKELEELEEELEQLRKELEELS-------RQISALRKDLARLEAEVEQLEERIAQLSKELTELEAEI--- 763
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 363 lkNKLNQDSGKSTTALHQENNKIKELSQEVERLKlklKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKM 442
Cdd:TIGR02168 764 --EELEERLEEAEEELAEAEAEIEELEAQIEQLK---EELKALREALDELRAELTLLNEEAANLRERLESLERRIAATER 838
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 443 ELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKEkihEYMATEDLICHLQGDHSVLQKKLNQQENRNRDLGR 522
Cdd:TIGR02168 839 RLEDLEEQIEELSEDIESLAAEIEELEELIEELESELEALLN---ERASLEEALALLRSELEELSEELRELESKRSELRR 915
|
570
....*....|...
gi 544346182 523 EIENLTKELERYR 535
Cdd:TIGR02168 916 ELEELREKLAQLE 928
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
4-280 |
9.51e-09 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 59.30 E-value: 9.51e-09
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQ--------------EEEQKATRLEKELQTQTTKFHQDQDTI 69
Cdd:TIGR02168 754 KELTELEAEIEELEERLEEAEEELAEAEAEIEELEAQIEqlkeelkalrealdELRAELTLLNEEAANLRERLESLERRI 833
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 70 MAK---LTNEDSQNRQLQQKLAALSRQIDELEEtnrSLRKAEEELQDIKEKISKGEygnagimaevEELRKRVLDMEGKD 146
Cdd:TIGR02168 834 AATerrLEDLEEQIEELSEDIESLAAEIEELEE---LIEELESELEALLNERASLE----------EALALLRSELEELS 900
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 147 EELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMAL-EKLEDAFNKSKQECYSLKCNLEKERMttkQLSQELESL 225
Cdd:TIGR02168 901 EELRELESKRSELRRELEELREKLAQLELRLEGLEVRIDNLqERLSEEYSLTLEEAEALENKIEDDEE---EARRRLKRL 977
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 226 KVRIKEL--------EAIESRLEKTEF--TLKEDLTK-LKTLTVMFVDERKTMSEKLKKTEDKLQA 280
Cdd:TIGR02168 978 ENKIKELgpvnlaaiEEYEELKERYDFltAQKEDLTEaKETLEEAIEEIDREARERFKDTFDQVNE 1043
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
16-443 |
1.47e-08 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 58.49 E-value: 1.47e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 16 QRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQID 95
Cdd:TIGR04523 181 EKLNIQKNIDKIKNKLLKLELLLSNLKKKIQKNKSLESQISELKKQNNQLKDNIEKKQQEINEKTTEISNTQTQLNQLKD 260
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 96 ELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKD--EELIKMEEQCRDLNKRLERETLQSKDF 173
Cdd:TIGR04523 261 EQNKIKKQLSEKQKELEQNNKKIKELEKQLNQLKSEISDLNNQKEQDWNKElkSELKNQEKKLEEIQNQISQNNKIISQL 340
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 174 KLEVEKLSKRIM------------------ALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAI 235
Cdd:TIGR04523 341 NEQISQLKKELTnsesensekqreleekqnEIEKLKKENQSYKQEIKNLESQINDLESKIQNQEKLNQQKDEQIKKLQQE 420
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 236 ESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLiEETKRALKSKTDvee 315
Cdd:TIGR04523 421 KELLEKEIERLKETIIKNNSEIKDLTNQDSVKELIIKNLDNTRESLETQLKVLSRSINKIKQNL-EQKQKELKSKEK--- 496
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 316 KMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRL----QSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIKELSQE 391
Cdd:TIGR04523 497 ELKKLNEEKKELEEKVKDLTKKISSLKEKIEKLESEKkekeSKISDLEDELNKDDFELKKENLEKEIDEKNKEIEELKQT 576
|
410 420 430 440 450
....*....|....*....|....*....|....*....|....*....|..
gi 544346182 392 VERLKlklKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKME 443
Cdd:TIGR04523 577 QKSLK---KKQEEKQELIDQKEKEKKDLIKEIEEKEKKISSLEKELEKAKKE 625
|
|
| rad50 |
TIGR00606 |
rad50; All proteins in this family for which functions are known are involvedin recombination, ... |
4-469 |
2.14e-08 |
|
rad50; All proteins in this family for which functions are known are involvedin recombination, recombinational repair, and/or non-homologous end joining.They are components of an exonuclease complex with MRE11 homologs. This family is distantly related to the SbcC family of bacterial proteins.This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University).
Pssm-ID: 129694 [Multi-domain] Cd Length: 1311 Bit Score: 58.13 E-value: 2.14e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEA---RVQEEEQKATRLEKEL-----QTQTTKFHQDQDTIMAKLTN 75
Cdd:TIGR00606 433 DEKKGLGRTIELKKEILEKKQEELKFVIKELQQLEGssdRILELDQELRKAERELskaekNSLTETLKKEVKSLQNEKAD 512
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 76 EDSQNRQLQQKLAALSRQIDELEETNRSLRK---AEEELQDIKEKISKGEYGNAGIMAEVEELRKRvldMEGKDEELIKM 152
Cdd:TIGR00606 513 LDRKLRKLDQEMEQLNHHTTTRTQMEMLTKDkmdKDEQIRKIKSRHSDELTSLLGYFPNKKQLEDW---LHSKSKEINQT 589
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 153 EEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALE-KLEDAFNKSKQECY--SLKCNLEKERMTTKQLSQELESLKVRI 229
Cdd:TIGR00606 590 RDRLAKLNKELASLEQNKNHINNELESKEEQLSSYEdKLFDVCGSQDEESDleRLKEEIEKSSKQRAMLAGATAVYSQFI 669
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 230 KELEAIES-------RLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEE 302
Cdd:TIGR00606 670 TQLTDENQsccpvcqRVFQTEAELQEFISDLQSKLRLAPDKLKSTESELKKKEKRRDEMLGLAPGRQSIIDLKEKEIPEL 749
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 303 TKRALKSKTDVEEKMYSVTKERDDLKNkLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQEN 382
Cdd:TIGR00606 750 RNKLQKVNRDIQRLKNDIEEQETLLGT-IMPEEESAKVCLTDVTIMERFQMELKDVERKIAQQAAKLQGSDLDRTVQQVN 828
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 383 NKIKE-------LSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAK----YKLAE 451
Cdd:TIGR00606 829 QEKQEkqheldtVVSKIELNRKLIQDQQEQIQHLKSKTNELKSEKLQIGTNLQRRQQFEEQLVELSTEVQSlireIKDAK 908
|
490
....*....|....*...
gi 544346182 452 KTETSHEQWLFKRLQEEE 469
Cdd:TIGR00606 909 EQDSPLETFLEKDQQEKE 926
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
11-541 |
2.16e-08 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 58.26 E-value: 2.16e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 11 AQLTLQRQKIQELTTNAKETHTKLALA-----------EARVQEEEQKATrlekELQTQTTKFHQDQDTIMAKLTNEDSQ 79
Cdd:pfam01576 43 NALQEQLQAETELCAEAEEMRARLAARkqeleeilhelESRLEEEEERSQ----QLQNEKKKMQQHIQDLEEQLDEEEAA 118
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 80 NRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELI-------KM 152
Cdd:pfam01576 119 RQKLQLEKVTTEAKIKKLEEDILLLEDQNSKLSKERKLLEERISEFTSNLAEEEEKAKSLSKLKNKHEAMIsdleerlKK 198
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 153 EEQCR----DLNKRLERETL----QSKDFKLEVEKLSKRIMALEK-LEDAFNKSKQECYSLKCNLEKERMTTKQLSQELE 223
Cdd:pfam01576 199 EEKGRqeleKAKRKLEGESTdlqeQIAELQAQIAELRAQLAKKEEeLQAALARLEEETAQKNNALKKIRELEAQISELQE 278
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 224 SLkvrikELE-AIESRLEKTEFTLKEDLTKLKTLTVMFVD----------ERKTMSEKLKKT-EDKLQAASSQLQVEQNK 291
Cdd:pfam01576 279 DL-----ESErAARNKAEKQRRDLGEELEALKTELEDTLDttaaqqelrsKREQEVTELKKAlEEETRSHEAQLQEMRQK 353
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 292 VTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEA--IEKDFLKNKLNQ 369
Cdd:pfam01576 354 HTQALEELTEQLEQAKRNKANLEKAKQALESENAELQAELRTLQQAKQDSEHKRKKLEGQLQELQArlSESERQRAELAE 433
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 370 DSGKS-------TTALHQENNKIKELSQEVERLKLKLKDMK-----------AIEDDLMKTEDEYETLERRYANERDKAQ 431
Cdd:pfam01576 434 KLSKLqselesvSSLLNEAEGKNIKLSKDVSSLESQLQDTQellqeetrqklNLSTRLRQLEDERNSLQEQLEEEEEAKR 513
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 432 FLSKELEHVKMELA--KYKLAEKTETSHeqwlfkrlQEEEAKSgHLSREVDALKEKIHEYMATEDlichlqgdhsvlqkK 509
Cdd:pfam01576 514 NVERQLSTLQAQLSdmKKKLEEDAGTLE--------ALEEGKK-RLQRELEALTQQLEEKAAAYD--------------K 570
|
570 580 590
....*....|....*....|....*....|..
gi 544346182 510 LNQQENRnrdLGREIENLTKELERYRHFSKSL 541
Cdd:pfam01576 571 LEKTKNR---LQQELDDLLVDLDHQRQLVSNL 599
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
90-417 |
2.55e-08 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 58.02 E-value: 2.55e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 90 LSRQIDELE------ETNRSLrKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRL 163
Cdd:COG1196 198 LERQLEPLErqaekaERYREL-KEELKELEAELLLLKLRELEAELEELEAELEELEAELEELEAELAELEAELEELRLEL 276
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 164 ERETLQSKDFKLEVEKLSKRIMALEKledafnkskqecyslkcNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTE 243
Cdd:COG1196 277 EELELELEEAQAEEYELLAELARLEQ-----------------DIARLEERRRELEERLEELEEELAELEEELEELEEEL 339
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 244 FTLKEDLTKLKTltvmfvdERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMysvTKE 323
Cdd:COG1196 340 EELEEELEEAEE-------ELEEAEAELAEAEEALLEAEAELAEAEEELEELAEELLEALRAAAELAAQLEELE---EAE 409
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 324 RDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLK-NKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDM 402
Cdd:COG1196 410 EALLERLERLEEELEELEEALAELEEEEEEEEEALEEAAEEeAELEEEEEALLELLAELLEEAALLEAALAELLEELAEA 489
|
330
....*....|....*
gi 544346182 403 KAIEDDLMKTEDEYE 417
Cdd:COG1196 490 AARLLLLLEAEADYE 504
|
|
| SMC_N |
pfam02463 |
RecF/RecN/SMC N terminal domain; This domain is found at the N terminus of SMC proteins. The ... |
4-337 |
5.81e-08 |
|
RecF/RecN/SMC N terminal domain; This domain is found at the N terminus of SMC proteins. The SMC (structural maintenance of chromosomes) superfamily proteins have ATP-binding domains at the N- and C-termini, and two extended coiled-coil domains separated by a hinge in the middle. The eukaryotic SMC proteins form two kind of heterodimers: the SMC1/SMC3 and the SMC2/SMC4 types. These heterodimers constitute an essential part of higher order complexes, which are involved in chromatin and DNA dynamics. This family also includes the RecF and RecN proteins that are involved in DNA metabolism and recombination.
Pssm-ID: 426784 [Multi-domain] Cd Length: 1161 Bit Score: 56.90 E-value: 5.81e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLE---KELQTQTTKFHQDQDTIMAKLTNEDSQN 80
Cdd:pfam02463 680 ELQEKAESELAKEEILRRQLEIKKKEQREKEELKKLKLEAEELLADRVQeaqDKINEELKLLKQKIDEEEEEEEKSRLKK 759
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 81 RQLQQKLAALSRQIDEL-EETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDL 159
Cdd:pfam02463 760 EEKEEEKSELSLKEKELaEEREKTEKLKVEEEKEEKLKAQEEELRALEEELKEEAELLEEEQLLIEQEEKIKEEELEELA 839
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 160 NKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRL 239
Cdd:pfam02463 840 LELKEEQKLEKLAEEELERLEEEITKEELLQELLLKEEELEEQKLKDELESKEEKEKEEKKELEEESQKLNLLEEKENEI 919
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 240 EKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYS 319
Cdd:pfam02463 920 EERIKEEAEILLKYEEEPEELLLEEADEKEKEENNKEEEEERNKRLLLAKEELGKVNLMAIEEFEEKEERYNKDELEKER 999
|
330
....*....|....*...
gi 544346182 320 VTKERDDLKNKLKAEEEK 337
Cdd:pfam02463 1000 LEEEKKKLIRAIIEETCQ 1017
|
|
| SCP-1 |
pfam05483 |
Synaptonemal complex protein 1 (SCP-1); Synaptonemal complex protein 1 (SCP-1) is the major ... |
6-531 |
8.96e-08 |
|
Synaptonemal complex protein 1 (SCP-1); Synaptonemal complex protein 1 (SCP-1) is the major component of the transverse filaments of the synaptonemal complex. Synaptonemal complexes are structures that are formed between homologous chromosomes during meiotic prophase.
Pssm-ID: 114219 [Multi-domain] Cd Length: 787 Bit Score: 55.88 E-value: 8.96e-08
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 6 QQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQ 85
Cdd:pfam05483 228 EEEYKKEINDKEKQVSLLLIQITEKENKMKDLTFLLEESRDKANQLEEKTKLQDENLKELIEKKDHLTKELEDIKMSLQR 307
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 86 KLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVldmegkdEELIKMEEQcrdlnkRLER 165
Cdd:pfam05483 308 SMSTQKALEEDLQIATKTICQLTEEKEAQMEELNKAKAAHSFVVTEFEATTCSL-------EELLRTEQQ------RLEK 374
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 166 ETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFT 245
Cdd:pfam05483 375 NEDQLKIITMELQKKSSELEEMTKFKNNKEVELEELKKILAEDEKLLDEKKQFEKIAEELKGKEQELIFLLQAREKEIHD 454
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 246 LKEDLTKLKTLTVMFVDERKTM-----SEKLKKTEdkLQAASSQLQVEQNKVTTVTEKLIEETK--------------RA 306
Cdd:pfam05483 455 LEIQLTAIKTSEEHYLKEVEDLkteleKEKLKNIE--LTAHCDKLLLENKELTQEASDMTLELKkhqediinckkqeeRM 532
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 307 LKSKTDVEEK-------MYSVTKE----RDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDsgKST 375
Cdd:pfam05483 533 LKQIENLEEKemnlrdeLESVREEfiqkGDEVKCKLDKSEENARSIEYEVLKKEKQMKILENKCNNLKKQIENKN--KNI 610
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 376 TALHQENNKIKELSQEverlklKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQflsKELEHVKmeLAKYKLAEKTET 455
Cdd:pfam05483 611 EELHQENKALKKKGSA------ENKQLNAYEIKVNKLELELASAKQKFEEIIDNYQ---KEIEDKK--ISEEKLLEEVEK 679
|
490 500 510 520 530 540 550
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 456 SH--EQWLFKRLQEEEAKSGHLSREVDALKEK-IHEYmatEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKEL 531
Cdd:pfam05483 680 AKaiADEAVKLQKEIDKRCQHKIAEMVALMEKhKHQY---DKIIEERDSELGLYKNKEQEQSSAKAALEIELSNIKAEL 755
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
4-369 |
1.16e-07 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 55.80 E-value: 1.16e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQE-EEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQ 82
Cdd:TIGR04523 260 DEQNKIKKQLSEKQKELEQNNKKIKELEKQLNQLKSEISDlNNQKEQDWNKELKSELKNQEKKLEEIQNQISQNNKIISQ 339
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 83 LQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKgeyGNAGIMAEVEELRKRVLDMEGK-----------DEELIK 151
Cdd:TIGR04523 340 LNEQISQLKKELTNSESENSEKQRELEEKQNEIEKLKK---ENQSYKQEIKNLESQINDLESKiqnqeklnqqkDEQIKK 416
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 152 MEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQ-----------ECYSLKCNLEKERMTTKQLSQ 220
Cdd:TIGR04523 417 LQQEKELLEKEIERLKETIIKNNSEIKDLTNQDSVKELIIKNLDNTREsletqlkvlsrSINKIKQNLEQKQKELKSKEK 496
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 221 ELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQaaSSQLQVEQNKVTTVTEKLI 300
Cdd:TIGR04523 497 ELKKLNEEKKELEEKVKDLTKKISSLKEKIEKLESEKKEKESKISDLEDELNKDDFELK--KENLEKEIDEKNKEIEELK 574
|
330 340 350 360 370 380 390
....*....|....*....|....*....|....*....|....*....|....*....|....*....|.
gi 544346182 301 EETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEK--DFLKNKLNQ 369
Cdd:TIGR04523 575 QTQKSLKKKQEEKQELIDQKEKEKKDLIKEIEEKEKKISSLEKELEKAKKENEKLSSIIKniKSKKNKLKQ 645
|
|
| MAD |
pfam05557 |
Mitotic checkpoint protein; This family consists of several eukaryotic mitotic checkpoint ... |
5-557 |
1.36e-07 |
|
Mitotic checkpoint protein; This family consists of several eukaryotic mitotic checkpoint (Mitotic arrest deficient or MAD) proteins. The mitotic spindle checkpoint monitors proper attachment of the bipolar spindle to the kinetochores of aligned sister chromatids and causes a cell cycle arrest in prometaphase when failures occur. Multiple components of the mitotic spindle checkpoint have been identified in yeast and higher eukaryotes. In S.cerevisiae, the existence of a Mad1-dependent complex containing Mad2, Mad3, Bub3 and Cdc20 has been demonstrated.
Pssm-ID: 461677 [Multi-domain] Cd Length: 660 Bit Score: 55.52 E-value: 1.36e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 5 EQQRLTAQLT-LQRQKIQ----------ELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKL 73
Cdd:pfam05557 3 ELIESKARLSqLQNEKKQmelehkrariELEKKASALKRQLDRESDRNQELQKRIRLLEKREAEAEEALREQAELNRLKK 82
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 74 TNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKME 153
Cdd:pfam05557 83 KYLEALNKKLNEKESQLADAREVISCLKNELSELRRQIQRAELELQSTNSELEELQERLDLLKAKASEAEQLRQNLEKQQ 162
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 154 EQCRDLNKR---LERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMttkqLSQELESLKVRIK 230
Cdd:pfam05557 163 SSLAEAEQRikeLEFEIQSQEQDSEIVKNSKSELARIPELEKELERLREHNKHLNENIENKLL----LKEEVEDLKRKLE 238
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 231 ELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMseklkKTEDKLQAASSQLQveQNKVTTVTEK--LIEETKRALK 308
Cdd:pfam05557 239 REEKYREEAATLELEKEKLEQELQSWVKLAQDTGLNL-----RSPEDLSRRIEQLQ--QREIVLKEENssLTSSARQLEK 311
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 309 SKTDVEEKMYSVTKERDDLKNKLKAEEEkgndllsrvnmLKNRLQ---SLEAIEKDFLKNKL-NQDSGKSTT-ALHQENN 383
Cdd:pfam05557 312 ARRELEQELAQYLKKIEDLNKKLKRHKA-----------LVRRLQrrvLLLTKERDGYRAILeSYDKELTMSnYSPQLLE 380
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 384 KIKELSQEVERLKLKLKDMKAieddlmktedEYETLERRYANERDKAQFLSKELEhvkmelakyklaektetsheqwlFK 463
Cdd:pfam05557 381 RIEEAEDMTQKMQAHNEEMEA----------QLSVAEEELGGYKQQAQTLERELQ-----------------------AL 427
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 464 RLQEEEAKSGHLSREVDALKEKIHEYMAT-----------EDLICH--LQGD-----HSVLQKKLNQQENRNRDLGREIE 525
Cdd:pfam05557 428 RQQESLADPSYSKEEVDSLRRKLETLELErqrlreqknelEMELERrcLQGDydpkkTKVLHLSMNPAAEAYQQRKNQLE 507
|
570 580 590
....*....|....*....|....*....|..
gi 544346182 526 NLTKELERYRHFSKSLRPSLNGRRISDPQVFS 557
Cdd:pfam05557 508 KLQAEIERLKRLLKKLEDDLEQVLRLPETTST 539
|
|
| COG5022 |
COG5022 |
Myosin heavy chain [General function prediction only]; |
78-597 |
2.37e-07 |
|
Myosin heavy chain [General function prediction only];
Pssm-ID: 227355 [Multi-domain] Cd Length: 1463 Bit Score: 55.08 E-value: 2.37e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 78 SQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGnagimaEVEELRKRVLDMEgKDEELIKMEEQCR 157
Cdd:COG5022 806 LGSRKEYRSYLACIIKLQKTIKREKKLRETEEVEFSLKAEVLIQKFG------RSLKAKKRFSLLK-KETIYLQSAQRVE 878
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 158 DLNKRLERETLQSKdfklEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTT-----------KQLSQELESLK 226
Cdd:COG5022 879 LAERQLQELKIDVK----SISSLKLVNLELESEIIELKKSLSSDLIENLEFKTELIARlkkllnnidleEGPSIEYVKLP 954
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 227 VRIKELEAiESRLEKTEFTLkEDLTKLKTLTVmfvDERKTMSEKLKKTEDKLQAASSQLQVEQNKVttvteKLIEETKRA 306
Cdd:COG5022 955 ELNKLHEV-ESKLKETSEEY-EDLLKKSTILV---REGNKANSELKNFKKELAELSKQYGALQEST-----KQLKELPVE 1024
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 307 LKSKTDVEEKMYSVTKErddLKNKLKAEEEKGNDLLSrVNMLKNRLQSLEaIEKDFLKNKLNQDSGKSTTalhqeNNKIK 386
Cdd:COG5022 1025 VAELQSASKIISSESTE---LSILKPLQKLKGLLLLE-NNQLQARYKALK-LRRENSLLDDKQLYQLEST-----ENLLK 1094
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 387 ElsqeverlkLKLKDMKAIEDDLMKTEDEYETL---ERRYANERDKAQFLSKELEHVKMELAKYKLAEKT---------- 453
Cdd:COG5022 1095 T---------INVKDLEVTNRNLVKPANVLQFIvaqMIKLNLLQEISKFLSQLVNTLEPVFQKLSVLQLEldglfweanl 1165
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 454 -ETSHEQWlFKRLQEEEAKSGH---------------LSREVDALKEKIHEYMATEDLICHL--QGDHSVLQKKLNQQEN 515
Cdd:COG5022 1166 eALPSPPP-FAALSEKRLYQSAlydeksklsssevndLKNELIALFSKIFSGWPRGDKLKKLisEGWVPTEYSTSLKGFN 1244
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 516 RNRDLGREIENLTKelERYRHFSKSLRPSLNGRRISdPQVFSKEVQTEAVDNEPPDYKSLIPLERAVINGQLYEESENQD 595
Cdd:COG5022 1245 NLNKKFDTPASMSN--EKLLSLLNSIDNLLSSYKLE-EEVLPATINSLLQYINVGLFNALRTKASSLRWKSATEVNYNSE 1321
|
..
gi 544346182 596 ED 597
Cdd:COG5022 1322 EL 1323
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
305-533 |
4.15e-07 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 54.17 E-value: 4.15e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 305 RALKSKTDVEEKMYSVtKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAiEKDFLKNKLNQDSGKSTTALHQENNK 384
Cdd:COG1196 216 RELKEELKELEAELLL-LKLRELEAELEELEAELEELEAELEELEAELAELEA-ELEELRLELEELELELEEAQAEEYEL 293
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 385 IKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETsheqwlfKR 464
Cdd:COG1196 294 LAELARLEQDIARLEERRRELEERLEELEEELAELEEELEELEEELEELEEELEEAEEELEEAEAELAEAE-------EA 366
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 465 LQEEEAKSGHLSREVDALKEKIHEymatedlichLQGDHSVLQKKLNQQENRNRDLGREIENLTKELER 533
Cdd:COG1196 367 LLEAEAELAEAEEELEELAEELLE----------ALRAAAELAAQLEELEEAEEALLERLERLEEELEE 425
|
|
| sbcc |
TIGR00618 |
exonuclease SbcC; All proteins in this family for which functions are known are part of an ... |
11-579 |
5.12e-07 |
|
exonuclease SbcC; All proteins in this family for which functions are known are part of an exonuclease complex with sbcD homologs. This complex is involved in the initiation of recombination to regulate the levels of palindromic sequences in DNA. This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University). [DNA metabolism, DNA replication, recombination, and repair]
Pssm-ID: 129705 [Multi-domain] Cd Length: 1042 Bit Score: 53.82 E-value: 5.12e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 11 AQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAAL 90
Cdd:TIGR00618 166 KELLMNLFPLDQYTQLALMEFAKKKSLHGKAELLTLRSQLLTLCTPCMPDTYHERKQVLEKELKHLREALQQTQQSHAYL 245
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 91 SRQIDELEETNRSLRKAEEELQDIKEKISKgeygnagiMAEVEELRKRvLDMEGKDEELIKMEEQCRDLNKRLER--ETL 168
Cdd:TIGR00618 246 TQKREAQEEQLKKQQLLKQLRARIEELRAQ--------EAVLEETQER-INRARKAAPLAAHIKAVTQIEQQAQRihTEL 316
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 169 QSKDFKLEVE-----KLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKvRIKELEAIESRLEKTE 243
Cdd:TIGR00618 317 QSKMRSRAKLlmkraAHVKQQSSIEEQRRLLQTLHSQEIHIRDAHEVATSIREISCQQHTLTQ-HIHTLQQQKTTLTQKL 395
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 244 FTLKEDLTKLKTL--TVMFVDERKtmseklKKTEDKLQAASSQLQVEQnKVTTVTEKLIEETKRALKSKTDVEEKMYSVT 321
Cdd:TIGR00618 396 QSLCKELDILQREqaTIDTRTSAF------RDLQGQLAHAKKQQELQQ-RYAELCAAAITCTAQCEKLEKIHLQESAQSL 468
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 322 KERD----DLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEaiekdflknklnqdsgKSTTALHQENNKIKELSQEVERLKL 397
Cdd:TIGR00618 469 KEREqqlqTKEQIHLQETRKKAVVLARLLELQEEPCPLC----------------GSCIHPNPARQDIDNPGPLTRRMQR 532
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 398 KLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSR 477
Cdd:TIGR00618 533 GEQTYAQLETSEEDVYHQLTSERKQRASLKEQMQEIQQSFSILTQCDNRSKEDIPNLQNITVRLQDLTEKLSEAEDMLAC 612
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 478 EVDALKEKIHEYMATEDLICHLQGDHSVLQKKLNQqenrnrdLGREIENLTKELERYRHFSKSLRPSLNG-RRISDPQVF 556
Cdd:TIGR00618 613 EQHALLRKLQPEQDLQDVRLHLQQCSQELALKLTA-------LHALQLTLTQERVREHALSIRVLPKELLaSRQLALQKM 685
|
570 580
....*....|....*....|...
gi 544346182 557 SKEVQTEAVDNEPPDYKSLIPLE 579
Cdd:TIGR00618 686 QSEKEQLTYWKEMLAQCQTLLRE 708
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
34-306 |
6.63e-07 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 52.46 E-value: 6.63e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 34 LALAEARVQEEEQKAtrLEKELQtqttKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEetnRSLRKAEEELQD 113
Cdd:COG4942 10 LLALAAAAQADAAAE--AEAELE----QLQQEIAELEKELAALKKEEKALLKQLAALERRIAALA---RRIRALEQELAA 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 114 IKEKISKGEygnagimAEVEELRKRvldmegKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDA 193
Cdd:COG4942 81 LEAELAELE-------KEIAELRAE------LEAQKEELAELLRALYRLGRQPPLALLLSPEDFLDAVRRLQYLKYLAPA 147
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 194 fnkskqecyslkcnlEKERMttKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLtvmfVDERKTMSEKLKK 273
Cdd:COG4942 148 ---------------RREQA--EELRADLAELAALRAELEAERAELEALLAELEEERAALEAL----KAERQKLLARLEK 206
|
250 260 270
....*....|....*....|....*....|...
gi 544346182 274 TEDKLQAASSQLQVEQNKVTTVTEKLIEETKRA 306
Cdd:COG4942 207 ELAELAAELAELQQEAEELEALIARLEAEAAAA 239
|
|
| PRK04778 |
PRK04778 |
septation ring formation regulator EzrA; Provisional |
33-537 |
7.05e-07 |
|
septation ring formation regulator EzrA; Provisional
Pssm-ID: 179877 [Multi-domain] Cd Length: 569 Bit Score: 52.92 E-value: 7.05e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 33 KLALAEARVQEEEQKATRLEKELQTQTT--KFHQDQDTIMAKltnedsqnrqlqqKLAALSRQIDELEETNRSLR--KAE 108
Cdd:PRK04778 38 KQELENLPVNDELEKVKKLNLTGQSEEKfeEWRQKWDEIVTN-------------SLPDIEEQLFEAEELNDKFRfrKAK 104
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 109 EELQDIKEKISKGEygnagimAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLeRETLQSKDFKL--EVEKLSKRima 186
Cdd:PRK04778 105 HEINEIESLLDLIE-------EDIEQILEELQELLESEEKNREEVEQLKDLYREL-RKSLLANRFSFgpALDELEKQ--- 173
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 187 LEKLEDAFNKSKQEcySLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTeftLKEDLTKLKT----------- 255
Cdd:PRK04778 174 LENLEEEFSQFVEL--TESGDYVEAREILDQLEEELAALEQIMEEIPELLKELQTE---LPDQLQELKAgyrelveegyh 248
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 256 LTVMFVDER-KTMSEKLKKTEDKLqaasSQLQVEqnkvttVTEKLIEETKRALKSKTDVEEKMY----SVTKERDDLKNK 330
Cdd:PRK04778 249 LDHLDIEKEiQDLKEQIDENLALL----EELDLD------EAEEKNEEIQERIDQLYDILEREVkarkYVEKNSDTLPDF 318
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 331 LKAEEEKGNDL-----------------LSRVNMLKNRLQSleaIEKDFLKNKLNQDSGKSTtalhqennkikeLSQEVE 393
Cdd:PRK04778 319 LEHAKEQNKELkeeidrvkqsytlneseLESVRQLEKQLES---LEKQYDEITERIAEQEIA------------YSELQE 383
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 394 RLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKL--------AEKTETSHE-QWLFKR 464
Cdd:PRK04778 384 ELEEILKQLEEIEKEQEKLSEMLQGLRKDELEAREKLERYRNKLHEIKRYLEKSNLpglpedylEMFFEVSDEiEALAEE 463
|
490 500 510 520 530 540 550
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*
gi 544346182 465 LQEEEAKSGHLSREVDALKEKIHE-YMATEDLIchlqgDHSVLQKKLNQQENRNRDLGREIEN-LTKELERYRHF 537
Cdd:PRK04778 464 LEEKPINMEAVNRLLEEATEDVETlEEETEELV-----ENATLTEQLIQYANRYRSDNEEVAEaLNEAERLFREY 533
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
17-165 |
7.77e-07 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 53.00 E-value: 7.77e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 17 RQKIQELTTnakethtKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDtiMAKLTNEDSQNRQLQQKLAALSRQIDE 96
Cdd:COG4913 609 RAKLAALEA-------ELAELEEELAEAEERLEALEAELDALQERREALQR--LAEYSWDEIDVASAEREIAELEAELER 679
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 97 LEETNRSLRKAEEELQDIKEKISKGEygnagimaevEELRKRVLDMEGKDEELIKMEEQCRDLNKRLER 165
Cdd:COG4913 680 LDASSDDLAALEEQLEELEAELEELE----------EELDELKGEIGRLEKELEQAEEELDELQDRLEA 738
|
|
| MAD |
pfam05557 |
Mitotic checkpoint protein; This family consists of several eukaryotic mitotic checkpoint ... |
6-442 |
8.33e-07 |
|
Mitotic checkpoint protein; This family consists of several eukaryotic mitotic checkpoint (Mitotic arrest deficient or MAD) proteins. The mitotic spindle checkpoint monitors proper attachment of the bipolar spindle to the kinetochores of aligned sister chromatids and causes a cell cycle arrest in prometaphase when failures occur. Multiple components of the mitotic spindle checkpoint have been identified in yeast and higher eukaryotes. In S.cerevisiae, the existence of a Mad1-dependent complex containing Mad2, Mad3, Bub3 and Cdc20 has been demonstrated.
Pssm-ID: 461677 [Multi-domain] Cd Length: 660 Bit Score: 52.82 E-value: 8.33e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 6 QQRLTAQLTLQRQKIQELTTNAKETHTKlalaEARVQEEEQKATRLEKELQTQTTKFHQDQdtimAKLTNEDSQNRQLQQ 85
Cdd:pfam05557 68 EEALREQAELNRLKKKYLEALNKKLNEK----ESQLADAREVISCLKNELSELRRQIQRAE----LELQSTNSELEELQE 139
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 86 KLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNA----------------GIMAEVEELRKRVLDMEGKDEEL 149
Cdd:pfam05557 140 RLDLLKAKASEAEQLRQNLEKQQSSLAEAEQRIKELEFEIQsqeqdseivknskselARIPELEKELERLREHNKHLNEN 219
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 150 IK----MEEQCRDLNKRLER-ETLQSKDFKLEVEK--------------------------LSKRIMALEKLEDAFnksK 198
Cdd:pfam05557 220 IEnkllLKEEVEDLKRKLEReEKYREEAATLELEKekleqelqswvklaqdtglnlrspedLSRRIEQLQQREIVL---K 296
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 199 QECYSLKCNLEKERMTTKQLSQE-------LESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKT----- 266
Cdd:pfam05557 297 EENSSLTSSARQLEKARRELEQElaqylkkIEDLNKKLKRHKALVRRLQRRVLLLTKERDGYRAILESYDKELTMsnysp 376
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 267 -MSEKLKKTEDKLQ-----AASSQLQVEQNKVTTVTEKLIEETK----RALKSKTDVEEKMYSvTKERDDLKNKLKAEEE 336
Cdd:pfam05557 377 qLLERIEEAEDMTQkmqahNEEMEAQLSVAEEELGGYKQQAQTLerelQALRQQESLADPSYS-KEEVDSLRRKLETLEL 455
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 337 KGNDLLSRVNMLKNRLQSLEaIEKDFLKNK---LNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTE 413
Cdd:pfam05557 456 ERQRLREQKNELEMELERRC-LQGDYDPKKtkvLHLSMNPAAEAYQQRKNQLEKLQAEIERLKRLLKKLEDDLEQVLRLP 534
|
490 500
....*....|....*....|....*....
gi 544346182 414 DEYETLERRYANERdKAQFLSKELEHVKM 442
Cdd:pfam05557 535 ETTSTMNFKEVLDL-RKELESAELKNQRL 562
|
|
| SMC_prok_B |
TIGR02168 |
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of ... |
207-546 |
8.57e-07 |
|
chromosome segregation protein SMC, common bacterial type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. This family represents the SMC protein of most bacteria. The smc gene is often associated with scpB (TIGR00281) and scpA genes, where scp stands for segregation and condensation protein. SMC was shown (in Caulobacter crescentus) to be induced early in S phase but present and bound to DNA throughout the cell cycle. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274008 [Multi-domain] Cd Length: 1179 Bit Score: 53.14 E-value: 8.57e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 207 NLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFtLKEDLTKL-KTLTVMFVDERKtmsEKLKKTEDKLQAASSQL 285
Cdd:TIGR02168 180 KLERTRENLDRLEDILNELERQLKSLERQAEKAERYKE-LKAELRELeLALLVLRLEELR---EELEELQEELKEAEEEL 255
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 286 QVEQNKVTTVTEKLieETKRALKSKtdVEEKMYSVTKERDDLKNKLkaeeekgNDLLSRVNMLKNRLQSLEaiekdflkN 365
Cdd:TIGR02168 256 EELTAELQELEEKL--EELRLEVSE--LEEEIEELQKELYALANEI-------SRLEQQKQILRERLANLE--------R 316
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 366 KLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANerdkaqfLSKELEHVKMELA 445
Cdd:TIGR02168 317 QLEELEAQLEELESKLDELAEELAELEEKLEELKEELESLEAELEELEAELEELESRLEE-------LEEQLETLRSKVA 389
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 446 KYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKEKIHEYMATEdlichLQGDHSVLQKKLNQQENRNRDLGREIE 525
Cdd:TIGR02168 390 QLELQIASLNNEIERLEARLERLEDRRERLQQEIEELLKKLEEAELKE-----LQAELEELEEELEELQEELERLEEALE 464
|
330 340
....*....|....*....|.
gi 544346182 526 NLTKELERYRHFSKSLRPSLN 546
Cdd:TIGR02168 465 ELREELEEAEQALDAAERELA 485
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
5-558 |
8.74e-07 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 52.72 E-value: 8.74e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 5 EQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQtqttKFHQDQDTIMAKLTNEDSQNRQLQ 84
Cdd:TIGR04523 83 QIKDLNDKLKKNKDKINKLNSDLSKINSEIKNDKEQKNKLEVELNKLEKQKK----ENKKNIDKFLTEIKKKEKELEKLN 158
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 85 QKLAALSRQIDELE----ETNRSLRKAEEELQDIKEKISKGEY----------GNAGIMAEVEELRKRVL---------- 140
Cdd:TIGR04523 159 NKYNDLKKQKEELEnelnLLEKEKLNIQKNIDKIKNKLLKLELllsnlkkkiqKNKSLESQISELKKQNNqlkdniekkq 238
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 141 -DMEGKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRImalEKLEDAFNKSKQECYSLkcNLEKERMTTKQLS 219
Cdd:TIGR04523 239 qEINEKTTEISNTQTQLNQLKDEQNKIKKQLSEKQKELEQNNKKI---KELEKQLNQLKSEISDL--NNQKEQDWNKELK 313
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 220 QELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQ-----------------AAS 282
Cdd:TIGR04523 314 SELKNQEKKLEEIQNQISQNNKIISQLNEQISQLKKELTNSESENSEKQRELEEKQNEIEklkkenqsykqeiknleSQI 393
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 283 SQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDF 362
Cdd:TIGR04523 394 NDLESKIQNQEKLNQQKDEQIKKLQQEKELLEKEIERLKETIIKNNSEIKDLTNQDSVKELIIKNLDNTRESLETQLKVL 473
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 363 LK--NKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKaqfLSKELEHV 440
Cdd:TIGR04523 474 SRsiNKIKQNLEQKQKELKSKEKELKKLNEEKKELEEKVKDLTKKISSLKEKIEKLESEKKEKESKISD---LEDELNKD 550
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 441 KMELAKYKLAEK--------TETSHEQWLFKRLQEE-EAKSGHLSREVDALKEKIHEYMATEdlichlqgdhSVLQKKLN 511
Cdd:TIGR04523 551 DFELKKENLEKEideknkeiEELKQTQKSLKKKQEEkQELIDQKEKEKKDLIKEIEEKEKKI----------SSLEKELE 620
|
570 580 590 600
....*....|....*....|....*....|....*....|....*..
gi 544346182 512 QQENRNRDLGREIENLTKELERYRHFSKSLRPSLNGRRISDPQVFSK 558
Cdd:TIGR04523 621 KAKKENEKLSSIIKNIKSKKNKLKQEVKQIKETIKEIRNKWPEIIKK 667
|
|
| PRK01156 |
PRK01156 |
chromosome segregation protein; Provisional |
2-552 |
9.02e-07 |
|
chromosome segregation protein; Provisional
Pssm-ID: 100796 [Multi-domain] Cd Length: 895 Bit Score: 52.98 E-value: 9.02e-07
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 2 VVDEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNR 81
Cdd:PRK01156 174 VIDMLRAEISNIDYLEEKLKSSNLELENIKKQIADDEKSHSITLKEIERLSIEYNNAMDDYNNLKSALNELSSLEDMKNR 253
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 82 qLQQKLAALSRQIDELEETNRSLRKAEEELQDIkekISKGEYGNAGIMAEVEELRKRVLD----MEGKDEELIKMEEQCR 157
Cdd:PRK01156 254 -YESEIKTAESDLSMELEKNNYYKELEERHMKI---INDPVYKNRNYINDYFKYKNDIENkkqiLSNIDAEINKYHAIIK 329
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 158 DLNKrLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQEL-ESLKVRIKELEAIE 236
Cdd:PRK01156 330 KLSV-LQKDYNDYIKKKSRYDDLNNQILELEGYEMDYNSYLKSIESLKKKIEEYSKNIERMSAFIsEILKIQEIDPDAIK 408
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 237 SRLEKTEFTLKEDLTKLKTLTVmfvdERKTMSEKLKKTEDKLQAASSQlqveqNKVTTVTEKLIEETKRALKSktDVEEK 316
Cdd:PRK01156 409 KELNEINVKLQDISSKVSSLNQ----RIRALRENLDELSRNMEMLNGQ-----SVCPVCGTTLGEEKSNHIIN--HYNEK 477
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 317 MYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLK-----------NRLQSLEAIEKDFlKNKLNQDSGKSTTAlHQENNKI 385
Cdd:PRK01156 478 KSRLEEKIREIEIEVKDIDEKIVDLKKRKEYLEseeinksineyNKIESARADLEDI-KIKINELKDKHDKY-EEIKNRY 555
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 386 KELSQEVERLKLK--LKDMKAIED-DLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLF 462
Cdd:PRK01156 556 KSLKLEDLDSKRTswLNALAVISLiDIETNRSRSNEIKKQLNDLESRLQEIEIGFPDDKSYIDKSIREIENEANNLNNKY 635
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 463 KRLQEeeaksghLSREVDALKEKIHEYMATEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKELERYRHFSKSLR 542
Cdd:PRK01156 636 NEIQE-------NKILIEKLRGKIDNYKKQIAEIDSIIPDLKEITSRINDIEDNLKKSRKALDDAKANRARLESTIEILR 708
|
570
....*....|..
gi 544346182 543 PSLN--GRRISD 552
Cdd:PRK01156 709 TRINelSDRIND 720
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
108-533 |
1.38e-06 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 52.08 E-value: 1.38e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 108 EEELQDIKEKISKGEYGNAGI-MAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMA 186
Cdd:COG4717 48 LERLEKEADELFKPQGRKPELnLKELKELEEELKEAEEKEEEYAELQEELEELEEELEELEAELEELREELEKLEKLLQL 127
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 187 LEKLEDafnkskqecyslkcnLEKERMTTKQLSQELESLKVRIKELEAIESRLEKteftLKEDLTKLKT-LTVMFVDERK 265
Cdd:COG4717 128 LPLYQE---------------LEALEAELAELPERLEELEERLEELRELEEELEE----LEAELAELQEeLEELLEQLSL 188
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 266 TMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLiEETKRALKSktdveekmYSVTKERDDLKNKLKAEEE--------- 336
Cdd:COG4717 189 ATEEELQDLAEELEELQQRLAELEEELEEAQEEL-EELEEELEQ--------LENELEAAALEERLKEARLllliaaall 259
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 337 ----KGNDLLSRVNMLKNRLQSLEAI------EKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIE 406
Cdd:COG4717 260 allgLGGSLLSLILTIAGVLFLVLGLlallflLLAREKASLGKEAEELQALPALEELEEEELEELLAALGLPPDLSPEEL 339
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 407 DDLMKTEDEYETLERRYANERDKAQFlsKELEHVKMELAKYKLAEkTETSHEQWL--FKRLQEEEAKSGHLSREVDALKE 484
Cdd:COG4717 340 LELLDRIEELQELLREAEELEEELQL--EELEQEIAALLAEAGVE-DEEELRAALeqAEEYQELKEELEELEEQLEELLG 416
|
410 420 430 440
....*....|....*....|....*....|....*....|....*....
gi 544346182 485 KIHEYMATEDLIcHLQGDHSVLQKKLNQQENRNRDLGREIENLTKELER 533
Cdd:COG4717 417 ELEELLEALDEE-ELEEELEELEEELEELEEELEELREELAELEAELEQ 464
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
231-527 |
2.86e-06 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 51.22 E-value: 2.86e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 231 ELEAIESRLEKTEFTLKEDLTKLKTLTvmfvDERKTmSEKLKKTEDKLQAASSQLQVEQNKVTtvtEKLIEETKRALKSK 310
Cdd:TIGR02169 178 ELEEVEENIERLDLIIDEKRQQLERLR----REREK-AERYQALLKEKREYEGYELLKEKEAL---ERQKEAIERQLASL 249
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 311 TDVEEKmysVTKERDDLKNKLKAEEEKGNDLLSRVNmlknRLQSLEAIEkdfLKNKLnqdsGKSTTALHQENNKIKELSQ 390
Cdd:TIGR02169 250 EEELEK---LTEEISELEKRLEEIEQLLEELNKKIK----DLGEEEQLR---VKEKI----GELEAEIASLERSIAEKER 315
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 391 EVERLKLKLKDmkaIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKyklaektetsheqwLFKRLQEEEA 470
Cdd:TIGR02169 316 ELEDAEERLAK---LEAEIDKLLAEIEELEREIEEERKRRDKLTEEYAELKEELED--------------LRAELEEVDK 378
|
250 260 270 280 290
....*....|....*....|....*....|....*....|....*....|....*..
gi 544346182 471 KSGHLSREVDALKEKIHEYmatEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENL 527
Cdd:TIGR02169 379 EFAETRDELKDYREKLEKL---KREINELKRELDRLQEELQRLSEELADLNAAIAGI 432
|
|
| rad50 |
TIGR00606 |
rad50; All proteins in this family for which functions are known are involvedin recombination, ... |
40-471 |
2.93e-06 |
|
rad50; All proteins in this family for which functions are known are involvedin recombination, recombinational repair, and/or non-homologous end joining.They are components of an exonuclease complex with MRE11 homologs. This family is distantly related to the SbcC family of bacterial proteins.This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University).
Pssm-ID: 129694 [Multi-domain] Cd Length: 1311 Bit Score: 51.20 E-value: 2.93e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 40 RVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKL----AALSRQIDELEETNRSLRKAEEELQDIK 115
Cdd:TIGR00606 685 RVFQTEAELQEFISDLQSKLRLAPDKLKSTESELKKKEKRRDEMLGLApgrqSIIDLKEKEIPELRNKLQKVNRDIQRLK 764
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 116 EKISKGEYGNAGIMAEvEELRKRVLDMEGKDEELikmEEQCRDLNKRLERET--LQSKDFKLEVEKLSKRIMALEKLEDA 193
Cdd:TIGR00606 765 NDIEEQETLLGTIMPE-EESAKVCLTDVTIMERF---QMELKDVERKIAQQAakLQGSDLDRTVQQVNQEKQEKQHELDT 840
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 194 FNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLtvmfVDERKTMSEKLKK 273
Cdd:TIGR00606 841 VVSKIELNRKLIQDQQEQIQHLKSKTNELKSEKLQIGTNLQRRQQFEEQLVELSTEVQSLIRE----IKDAKEQDSPLET 916
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 274 TEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKE-RDDLKNKLKAEEEKGNDLLSRVNMLKNRL 352
Cdd:TIGR00606 917 FLEKDQQEKEELISSKETSNKKAQDKVNDIKEKVKNIHGYMKDIENKIQDgKDDYLKQKETELNTVNAQLEECEKHQEKI 996
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 353 -QSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIE--DDLMKTEDEYETLERRYANERDK 429
Cdd:TIGR00606 997 nEDMRLMRQDIDTQKIQERWLQDNLTLRKRENELKEVEEELKQHLKEMGQMQVLQmkQEHQKLEENIDLIKRNHVLALGR 1076
|
410 420 430 440
....*....|....*....|....*....|....*....|..
gi 544346182 430 AQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAK 471
Cdd:TIGR00606 1077 QKGYEKEIKHFKKELREPQFRDAEEKYREMMIVMRTTELVNK 1118
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
42-531 |
3.25e-06 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 50.94 E-value: 3.25e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 42 QEEEQKATRLE----KELQTQTTKFHQDQDTIMAKLTNEDSQnrqLQQKLAALSRQIDELEETNRSL--RKAEEE--LQD 113
Cdd:pfam01576 3 QEEEMQAKEEElqkvKERQQKAESELKELEKKHQQLCEEKNA---LQEQLQAETELCAEAEEMRARLaaRKQELEeiLHE 79
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 114 IKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEElikmEEQCRDlNKRLERETLQSKdfkleVEKLSKRIMALEKLEDA 193
Cdd:pfam01576 80 LESRLEEEEERSQQLQNEKKKMQQHIQDLEEQLDE----EEAARQ-KLQLEKVTTEAK-----IKKLEEDILLLEDQNSK 149
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 194 FNKSK----QECYSLKCNLEKERMTTKQLSQ-------ELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVD 262
Cdd:pfam01576 150 LSKERklleERISEFTSNLAEEEEKAKSLSKlknkheaMISDLEERLKKEEKGRQELEKAKRKLEGESTDLQEQIAELQA 229
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 263 ERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVE-EKMY--SVTKERDDLKNKLKAEEEKGN 339
Cdd:pfam01576 230 QIAELRAQLAKKEEELQAALARLEEETAQKNNALKKIRELEAQISELQEDLEsERAArnKAEKQRRDLGEELEALKTELE 309
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 340 DLLSRVNMlKNRLQSLEAIEKDFLKNKLnQDSGKSTTALHQE-----NNKIKELSQEVE---RLKLKL-KDMKAIEDDLM 410
Cdd:pfam01576 310 DTLDTTAA-QQELRSKREQEVTELKKAL-EEETRSHEAQLQEmrqkhTQALEELTEQLEqakRNKANLeKAKQALESENA 387
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 411 KTEDEYETL-----ERRYANERDKAQFLSKELEHVKMELAKYKLAEKTE--TSHEQWLFKRLQEEEAKSGHLSREVDALK 483
Cdd:pfam01576 388 ELQAELRTLqqakqDSEHKRKKLEGQLQELQARLSESERQRAELAEKLSklQSELESVSSLLNEAEGKNIKLSKDVSSLE 467
|
490 500 510 520 530
....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 484 EKIHEY-----------MATEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKEL 531
Cdd:pfam01576 468 SQLQDTqellqeetrqkLNLSTRLRQLEDERNSLQEQLEEEEEAKRNVERQLSTLQAQL 526
|
|
| COG2433 |
COG2433 |
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only]; |
44-194 |
4.92e-06 |
|
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only];
Pssm-ID: 441980 [Multi-domain] Cd Length: 644 Bit Score: 50.24 E-value: 4.92e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 44 EEQKATRLEKELQT-QTTKFHQDQDtimakLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGE 122
Cdd:COG2433 383 EELIEKELPEEEPEaEREKEHEERE-----LTEEEEEIRRLEEQVERLEAEVEELEAELEEKDERIERLERELSEARSEE 457
|
90 100 110 120 130 140 150
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 123 YGNAGIMAEVEELRKRVLDMEGkdeELIKMEEQCRDLNKRLER-ETLQSKDFK------LEVEKLSKRimALEKLEDAF 194
Cdd:COG2433 458 RREIRKDREISRLDREIERLER---ELEEERERIEELKRKLERlKELWKLEHSgelvpvKVVEKFTKE--AIRRLEEEY 531
|
|
| EzrA |
pfam06160 |
Septation ring formation regulator, EzrA; During the bacterial cell cycle, the tubulin-like ... |
70-422 |
8.91e-06 |
|
Septation ring formation regulator, EzrA; During the bacterial cell cycle, the tubulin-like cell-division protein FtsZ polymerizes into a ring structure that establishes the location of the nascent division site. EzrA modulates the frequency and position of FtsZ ring formation. The structure contains 5 spectrin like alpha helical repeats.
Pssm-ID: 428797 [Multi-domain] Cd Length: 542 Bit Score: 49.47 E-value: 8.91e-06
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 70 MAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISK------GEYGNAgimaeVEELRKRVLDME 143
Cdd:pfam06160 85 KKALDEIEELLDDIEEDIKQILEELDELLESEEKNREEVEELKDKYRELRKtllanrFSYGPA-----IDELEKQLAEIE 159
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 144 GKDEEL---------IKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRI-MALEKLEDAFNKSKQECYSLK-CNLEKER 212
Cdd:pfam06160 160 EEFSQFeeltesgdyLEAREVLEKLEEETDALEELMEDIPPLYEELKTELpDQLEELKEGYREMEEEGYALEhLNVDKEI 239
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 213 mttKQLSQELESLKVRIKELEaiesrLEKTEFTLKEDLTKLKTLTVMF---VDERKTMSEKLKKTEDKLQAASSQLQVEQ 289
Cdd:pfam06160 240 ---QQLEEQLEENLALLENLE-----LDEAEEALEEIEERIDQLYDLLekeVDAKKYVEKNLPEIEDYLEHAEEQNKELK 311
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 290 NKVTTVTEKLI---EETKRALKSKTDVE--EKMYSVTKERDD--------LKNKLKAEEEKGNDLLSRVNMLKNRLQSLE 356
Cdd:pfam06160 312 EELERVQQSYTlneNELERVRGLEKQLEelEKRYDEIVERLEekevayseLQEELEEILEQLEEIEEEQEEFKESLQSLR 391
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 357 AIEKDFLKNKLNQDSGKSTTALHQEN------------------NKIKELSQEVERLKLklkDMKAIEDDLMKTEDEYET 418
Cdd:pfam06160 392 KDELEAREKLDEFKLELREIKRLVEKsnlpglpesyldyffdvsDEIEDLADELNEVPL---NMDEVNRLLDEAQDDVDT 468
|
....
gi 544346182 419 LERR 422
Cdd:pfam06160 469 LYEK 472
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
116-445 |
1.10e-05 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 49.24 E-value: 1.10e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 116 EKISKGEYGNAGIMAEVEELR-----KRVLDMEGKDEELI----KMEEQCRDLNKRLE-RETLQSKDFKLEVE----KLS 181
Cdd:COG3206 71 SGLSSLSASDSPLETQIEILKsrpvlERVVDKLNLDEDPLgeeaSREAAIERLRKNLTvEPVKGSNVIEISYTspdpELA 150
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 182 KRImaLEKLEDAFNKskqecYSLKCNLEKERMTTKQLSQELESLKvriKELEAIESRLEktEFTLKEDLTKLKtltvmfv 261
Cdd:COG3206 151 AAV--ANALAEAYLE-----QNLELRREEARKALEFLEEQLPELR---KELEEAEAALE--EFRQKNGLVDLS------- 211
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 262 DERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLieETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDL 341
Cdd:COG3206 212 EEAKLLLQQLSELESQLAEARAELAEAEARLAALRAQL--GSGPDALPELLQSPVIQQLRAQLAELEAELAELSARYTPN 289
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 342 LSRVNMLKNRLQSLEAiekdflknKLNQDSGKSTTALHQEnnkIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLER 421
Cdd:COG3206 290 HPDVIALRAQIAALRA--------QLQQEAQRILASLEAE---LEALQAREASLQAQLAQLEARLAELPELEAELRRLER 358
|
330 340
....*....|....*....|....
gi 544346182 422 RYANERDKAQFLSKELEHVKMELA 445
Cdd:COG3206 359 EVEVARELYESLLQRLEEARLAEA 382
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
5-488 |
1.12e-05 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 49.40 E-value: 1.12e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 5 EQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEK---ELQTQTTKFHQDQDTIMAKLTNEDSQNR 81
Cdd:pfam01576 216 ESTDLQEQIAELQAQIAELRAQLAKKEEELQAALARLEEETAQKNNALKkirELEAQISELQEDLESERAARNKAEKQRR 295
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 82 QLQQKLAALSRQI-DELEETNrslrkAEEELQDIKEkiskgeygnagimAEVEELRKRVldmegkDEELIKMEEQCRDLN 160
Cdd:pfam01576 296 DLGEELEALKTELeDTLDTTA-----AQQELRSKRE-------------QEVTELKKAL------EEETRSHEAQLQEMR 351
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 161 KRlERETLQSKDFKLEVEKLSKriMALEKLEDAFNKSKQEcyslkcnLEKERMTTKQLSQELE----SLKVRIKELEAIE 236
Cdd:pfam01576 352 QK-HTQALEELTEQLEQAKRNK--ANLEKAKQALESENAE-------LQAELRTLQQAKQDSEhkrkKLEGQLQELQARL 421
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 237 SRLEKTEFTLKEDLTKLKTltvmfvdERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTdveeK 316
Cdd:pfam01576 422 SESERQRAELAEKLSKLQS-------ELESVSSLLNEAEGKNIKLSKDVSSLESQLQDTQELLQEETRQKLNLST----R 490
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 317 MYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSleaiekdfLKNKLNQDSGksttALHQENNKIKELSQEVERLK 396
Cdd:pfam01576 491 LRQLEDERNSLQEQLEEEEEAKRNVERQLSTLQAQLSD--------MKKKLEEDAG----TLEALEEGKKRLQRELEALT 558
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 397 LKLKDMKAIEDDLMKT----EDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKS 472
Cdd:pfam01576 559 QQLEEKAAAYDKLEKTknrlQQELDDLLVDLDHQRQLVSNLEKKQKKFDQMLAEEKAISARYAEERDRAEAEAREKETRA 638
|
490
....*....|....*.
gi 544346182 473 GHLSREVDALKEKIHE 488
Cdd:pfam01576 639 LSLARALEEALEAKEE 654
|
|
| CwlO1 |
COG3883 |
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function ... |
72-291 |
1.15e-05 |
|
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function unknown];
Pssm-ID: 443091 [Multi-domain] Cd Length: 379 Bit Score: 48.67 E-value: 1.15e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 72 KLTNEDSQNRQLQQKLAALSRQID----ELEETNRSLRKAEEELQDIKEKISKGEygnagimAEVEELRKRVldmegkDE 147
Cdd:COG3883 17 QIQAKQKELSELQAELEAAQAELDalqaELEELNEEYNELQAELEALQAEIDKLQ-------AEIAEAEAEI------EE 83
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 148 ELIKMEEQCRDLNKRLERET-----LQSKDFklevEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERmttKQLSQEL 222
Cdd:COG3883 84 RREELGERARALYRSGGSVSyldvlLGSESF----SDFLDRLSALSKIADADADLLEELKADKAELEAKK---AELEAKL 156
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 223 ESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNK 291
Cdd:COG3883 157 AELEALKAELEAAKAELEAQQAEQEALLAQLSAEEAAAEAQLAELEAELAAAEAAAAAAAAAAAAAAAA 225
|
|
| 46 |
PHA02562 |
endonuclease subunit; Provisional |
33-272 |
1.29e-05 |
|
endonuclease subunit; Provisional
Pssm-ID: 222878 [Multi-domain] Cd Length: 562 Bit Score: 48.86 E-value: 1.29e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 33 KLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQ-QKLAALSRQID-ELEETNRSLRKAEEE 110
Cdd:PHA02562 170 KLNKDKIRELNQQIQTLDMKIDHIQQQIKTYNKNIEEQRKKNGENIARKQNKyDELVEEAKTIKaEIEELTDELLNLVMD 249
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 111 LQDIKEKISKGEYGNAGIMAEVEELRKrVLDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKL 190
Cdd:PHA02562 250 IEDPSAALNKLNTAAAKIKSKIEQFQK-VIKMYEKGGVCPTCTQQISEGPDRITKIKDKLKELQHSLEKLDTAIDELEEI 328
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 191 EDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAiESRLEKTEF-TLKEDLTKL---KTLTVMFVDERKT 266
Cdd:PHA02562 329 MDEFNEQSKKLLELKNKISTNKQSLITLVDKAKKVKAAIEELQA-EFVDNAEELaKLQDELDKIvktKSELVKEKYHRGI 407
|
....*.
gi 544346182 267 MSEKLK 272
Cdd:PHA02562 408 VTDLLK 413
|
|
| PTZ00121 |
PTZ00121 |
MAEBL; Provisional |
27-413 |
1.67e-05 |
|
MAEBL; Provisional
Pssm-ID: 173412 [Multi-domain] Cd Length: 2084 Bit Score: 48.98 E-value: 1.67e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 27 AKETHTKLALAEARVQEEEQKATRLEK--------ELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELE 98
Cdd:PTZ00121 1527 AKKAEEAKKADEAKKAEEKKKADELKKaeelkkaeEKKKAEEAKKAEEDKNMALRKAEEAKKAEEARIEEVMKLYEEEKK 1606
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 99 ETNRSLRKAEEE---------LQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQcrdlnkrlerETLQ 169
Cdd:PTZ00121 1607 MKAEEAKKAEEAkikaeelkkAEEEKKKVEQLKKKEAEEKKKAEELKKAEEENKIKAAEEAKKAEE----------DKKK 1676
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 170 SKDFKLEVEKLSKRIMALEKLEDAFNKSKQecysLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKED 249
Cdd:PTZ00121 1677 AEEAKKAEEDEKKAAEALKKEAEEAKKAEE----LKKKEAEEKKKAEELKKAEEENKIKAEEAKKEAEEDKKKAEEAKKD 1752
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 250 LTKLKTLTVMFVDERKTMSEKLKKTEdklqaassqlQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKN 329
Cdd:PTZ00121 1753 EEEKKKIAHLKKEEEKKAEEIRKEKE----------AVIEEELDEEDEKRRMEVDKKIKDIFDNFANIIEGGKEGNLVIN 1822
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 330 KLK-AEEEKGNDLLSRVNMLKNRLQSLEaiEKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVErlklKLKDMKAIEDD 408
Cdd:PTZ00121 1823 DSKeMEDSAIKEVADSKNMQLEEADAFE--KHKFNKNNENGEDGNKEADFNKEKDLKEDDEEEIE----EADEIEKIDKD 1896
|
....*
gi 544346182 409 LMKTE 413
Cdd:PTZ00121 1897 DIERE 1901
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
4-241 |
1.77e-05 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 47.84 E-value: 1.77e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQL 83
Cdd:COG4942 20 DAAAEAEAELEQLQQEIAELEKELAALKKEEKALLKQLAALERRIAALARRIRALEQELAALEAELAELEKEIAELRAEL 99
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 84 QQKLAALSRQIDELEetnRSLRKAEEEL----QDIKEKISKGEYgnagIMAEVEELRKRVLDMEGKDEELikmeeqcRDL 159
Cdd:COG4942 100 EAQKEELAELLRALY---RLGRQPPLALllspEDFLDAVRRLQY----LKYLAPARREQAEELRADLAEL-------AAL 165
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 160 NKRLERETLQSKDFKLEVEKlskrimALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRL 239
Cdd:COG4942 166 RAELEAERAELEALLAELEE------ERAALEALKAERQKLLARLEKELAELAAELAELQQEAEELEALIARLEAEAAAA 239
|
..
gi 544346182 240 EK 241
Cdd:COG4942 240 AE 241
|
|
| rad50 |
TIGR00606 |
rad50; All proteins in this family for which functions are known are involvedin recombination, ... |
2-644 |
2.08e-05 |
|
rad50; All proteins in this family for which functions are known are involvedin recombination, recombinational repair, and/or non-homologous end joining.They are components of an exonuclease complex with MRE11 homologs. This family is distantly related to the SbcC family of bacterial proteins.This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University).
Pssm-ID: 129694 [Multi-domain] Cd Length: 1311 Bit Score: 48.50 E-value: 2.08e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 2 VVDEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLE----------KELQTQTTKFHQDQDTIMA 71
Cdd:TIGR00606 194 VRQTQGQKVQEHQMELKYLKQYKEKACEIRDQITSKEAQLESSREIVKSYEneldplknrlKEIEHNLSKIMKLDNEIKA 273
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 72 KLTNE---DSQNRQLQQKLA----ALSRQIDELEETN-RSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRV---- 139
Cdd:TIGR00606 274 LKSRKkqmEKDNSELELKMEkvfqGTDEQLNDLYHNHqRTVREKERELVDCQRELEKLNKERRLLNQEKTELLVEQgrlq 353
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 140 LDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFkLEVEKLSKRIMALEKLEDAFNKSKQECYSLKcnlEKERMTTKQLS 219
Cdd:TIGR00606 354 LQADRHQEHIRARDSLIQSLATRLELDGFERGPF-SERQIKNFHTLVIERQEDEAKTAAQLCADLQ---SKERLKQEQAD 429
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 220 QELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMfVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKL 299
Cdd:TIGR00606 430 EIRDEKKGLGRTIELKKEILEKKQEELKFVIKELQQLEGS-SDRILELDQELRKAERELSKAEKNSLTETLKKEVKSLQN 508
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 300 ----IEETKRALKSK-------TDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRV-------NMLKNRLQSLEA---I 358
Cdd:TIGR00606 509 ekadLDRKLRKLDQEmeqlnhhTTTRTQMEMLTKDKMDKDEQIRKIKSRHSDELTSLlgyfpnkKQLEDWLHSKSKeinQ 588
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 359 EKDFLKnKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDM---KAIEDDLMKTEDEYETLERRYANERDKAQFLSK 435
Cdd:TIGR00606 589 TRDRLA-KLNKELASLEQNKNHINNELESKEEQLSSYEDKLFDVcgsQDEESDLERLKEEIEKSSKQRAMLAGATAVYSQ 667
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 436 ELEHVKME------LAKYKLAEKTETsheQWLFKRLQEEEAKSGHLSREVDALKEKIHEymATEDLICHLQGDHSVLQ-- 507
Cdd:TIGR00606 668 FITQLTDEnqsccpVCQRVFQTEAEL---QEFISDLQSKLRLAPDKLKSTESELKKKEK--RRDEMLGLAPGRQSIIDlk 742
|
570 580 590 600 610 620 630 640
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 508 -KKLNQQENRNRDLGREIENLTKELERYRHFSKSLRPSLNGRRISDPQVFSKEVQTEAVDNEPPDYKSLIPLERAVINGQ 586
Cdd:TIGR00606 743 eKEIPELRNKLQKVNRDIQRLKNDIEEQETLLGTIMPEEESAKVCLTDVTIMERFQMELKDVERKIAQQAAKLQGSDLDR 822
|
650 660 670 680 690
....*....|....*....|....*....|....*....|....*....|....*...
gi 544346182 587 LYEESENQDEDPNDEGSVLSFKCSQSTPCPVNRKLWIPWMKSKEGHLQNGKMQTKPNA 644
Cdd:TIGR00606 823 TVQQVNQEKQEKQHELDTVVSKIELNRKLIQDQQEQIQHLKSKTNELKSEKLQIGTNL 880
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
7-169 |
2.40e-05 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 46.84 E-value: 2.40e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 7 QRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNrqLQQK 86
Cdd:COG1579 20 DRLEHRLKELPAELAELEDELAALEARLEAAKTELEDLEKEIKRLELEIEEVEARIKKYEEQLGNVRNNKEYEA--LQKE 97
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 87 LAALSRQIDELEEtnrSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRV-LDMEGKDEELIKMEEQCRDLNKRLER 165
Cdd:COG1579 98 IESLKRRISDLED---EILELMERIEELEEELAELEAELAELEAELEEKKAELdEELAELEAELEELEAEREELAAKIPP 174
|
....
gi 544346182 166 ETLQ 169
Cdd:COG1579 175 ELLA 178
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
5-438 |
2.61e-05 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 48.01 E-value: 2.61e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 5 EQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQttkfhQDQDTIMAKLTNEDSQNRQLQ 84
Cdd:COG1196 406 EEAEEALLERLERLEEELEELEEALAELEEEEEEEEEALEEAAEEEAELEEEEE-----ALLELLAELLEEAALLEAALA 480
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 85 QKLAALSRQIDELEETNRSLRKAEEELQDIKEKisKGEYGNAGIMAEVEELRKRVLDMEGKDEELIkmEEQCRDLNKRLE 164
Cdd:COG1196 481 ELLEELAEAAARLLLLLEAEADYEGFLEGVKAA--LLLAGLRGLAGAVAVLIGVEAAYEAALEAAL--AAALQNIVVEDD 556
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 165 RETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQEcysLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEF 244
Cdd:COG1196 557 EVAAAAIEYLKAAKAGRATFLPLDKIRARAALAAAL---ARGAIGAAVDLVASDLREADARYYVLGDTLLGRTLVAARLE 633
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 245 TLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKmysvTKER 324
Cdd:COG1196 634 AALRRAVTLAGRLREVTLEGEGGSAGGSLTGGSRRELLAALLEAEAELEELAERLAEEELELEEALLAEEEE----EREL 709
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 325 DDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDsgksttalHQENNKIKELSQEVERLKLKLKDMKA 404
Cdd:COG1196 710 AEAEEERLEEELEEEALEEQLEAEREELLEELLEEEELLEEEALEE--------LPEPPDLEELERELERLEREIEALGP 781
|
410 420 430
....*....|....*....|....*....|....
gi 544346182 405 IEddlMKTEDEYETLERRYanerdkaQFLSKELE 438
Cdd:COG1196 782 VN---LLAIEEYEELEERY-------DFLSEQRE 805
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
4-533 |
2.66e-05 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 47.99 E-value: 2.66e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAE-ARVQEEEQKATRLEKELQTQTTKFHQ-DQDTIMAKLTNEDS--- 78
Cdd:COG4913 302 AELARLEAELERLEARLDALREELDELEAQIRGNGgDRLEQLEREIERLERELEERERRRARlEALLAALGLPLPASaee 381
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 79 ---QNRQLQQKLAALSRQIDELE----ETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDE---- 147
Cdd:COG4913 382 faaLRAEAAALLEALEEELEALEealaEAEAALRDLRRELRELEAEIASLERRKSNIPARLLALRDALAEALGLDEaelp 461
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 148 ---ELIKMEEQCRDLNKRLEReTLQSKDFKLEVEklskrimalEKLEDAFNKsKQECYSLKCNLEKERMTTKQLSQELES 224
Cdd:COG4913 462 fvgELIEVRPEEERWRGAIER-VLGGFALTLLVP---------PEHYAAALR-WVNRLHLRGRLVYERVRTGLPDPERPR 530
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 225 L-------KVRIKELEA---IESRLEKTEFTLK-EDLTKLK------TLTVMfVDERKTMSEK----------------- 270
Cdd:COG4913 531 LdpdslagKLDFKPHPFrawLEAELGRRFDYVCvDSPEELRrhpraiTRAGQ-VKGNGTRHEKddrrrirsryvlgfdnr 609
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 271 ---------LKKTEDKLQAASSQLQVEQNKVTTVTEKLieETKRALKSKTDVEEKMYSVTKERDDLKNKLkAEEEKGNDL 341
Cdd:COG4913 610 aklaaleaeLAELEEELAEAEERLEALEAELDALQERR--EALQRLAEYSWDEIDVASAEREIAELEAEL-ERLDASSDD 686
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 342 LSRvnmLKNRLQSLEAIEKDflknkLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDdlmktEDEYETLER 421
Cdd:COG4913 687 LAA---LEEQLEELEAELEE-----LEEELDELKGEIGRLEKELEQAEEELDELQDRLEAAEDLAR-----LELRALLEE 753
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 422 RYAN------ERDKAQFLSKELEHVKMELAkyKLAEKTETSHEQwlFKRLQEEEAksGHLSREVDALKE--KIHEYMATE 493
Cdd:COG4913 754 RFAAalgdavERELRENLEERIDALRARLN--RAEEELERAMRA--FNREWPAET--ADLDADLESLPEylALLDRLEED 827
|
570 580 590 600
....*....|....*....|....*....|....*....|
gi 544346182 494 DLICHlqgdhsvlQKKLNQQENRNrdLGREIENLTKELER 533
Cdd:COG4913 828 GLPEY--------EERFKELLNEN--SIEFVADLLSKLRR 857
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
352-535 |
3.01e-05 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 47.84 E-value: 3.01e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 352 LQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANERDKAQ 431
Cdd:COG4717 40 LAFIRAMLLERLEKEADELFKPQGRKPELNLKELKELEEELKEAEEKEEEYAELQEELEELEEELEELEAELEELREELE 119
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 432 FLSKELEHVKMELAKYKLAEKTETSHEQWlfKRLQEEEAKSGHLSREVDALKEKIHEY------------MATEDLICHL 499
Cdd:COG4717 120 KLEKLLQLLPLYQELEALEAELAELPERL--EELEERLEELRELEEELEELEAELAELqeeleelleqlsLATEEELQDL 197
|
170 180 190
....*....|....*....|....*....|....*.
gi 544346182 500 QGDHSVLQKKLNQQENRNRDLGREIENLTKELERYR 535
Cdd:COG4717 198 AEELEELQQRLAELEEELEEAQEELEELEEELEQLE 233
|
|
| PRK01156 |
PRK01156 |
chromosome segregation protein; Provisional |
40-360 |
3.22e-05 |
|
chromosome segregation protein; Provisional
Pssm-ID: 100796 [Multi-domain] Cd Length: 895 Bit Score: 47.97 E-value: 3.22e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 40 RVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEE---------- 109
Cdd:PRK01156 378 KIEEYSKNIERMSAFISEILKIQEIDPDAIKKELNEINVKLQDISSKVSSLNQRIRALRENLDELSRNMEmlngqsvcpv 457
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 110 ---ELQDIKEKISKGEYGN--AGIMAEVEELRKRVLDMEGKDEELIKMEE--QCRDLNkRLERETLQSKDFKLEVEKLSK 182
Cdd:PRK01156 458 cgtTLGEEKSNHIINHYNEkkSRLEEKIREIEIEVKDIDEKIVDLKKRKEylESEEIN-KSINEYNKIESARADLEDIKI 536
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 183 RIMALEKLEDAFNKSKQECYSLKCNLEKERMTT--KQLSQ----ELESLKVRI----KELEAIESRLEKTEFTLKEDLTK 252
Cdd:PRK01156 537 KINELKDKHDKYEEIKNRYKSLKLEDLDSKRTSwlNALAVisliDIETNRSRSneikKQLNDLESRLQEIEIGFPDDKSY 616
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 253 LKTLTVMFVDE------RKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTD---VEEKMYSVTKE 323
Cdd:PRK01156 617 IDKSIREIENEannlnnKYNEIQENKILIEKLRGKIDNYKKQIAEIDSIIPDLKEITSRINDIEDNlkkSRKALDDAKAN 696
|
330 340 350
....*....|....*....|....*....|....*..
gi 544346182 324 RDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEK 360
Cdd:PRK01156 697 RARLESTIEILRTRINELSDRINDINETLESMKKIKK 733
|
|
| SMC_N |
pfam02463 |
RecF/RecN/SMC N terminal domain; This domain is found at the N terminus of SMC proteins. The ... |
186-542 |
4.01e-05 |
|
RecF/RecN/SMC N terminal domain; This domain is found at the N terminus of SMC proteins. The SMC (structural maintenance of chromosomes) superfamily proteins have ATP-binding domains at the N- and C-termini, and two extended coiled-coil domains separated by a hinge in the middle. The eukaryotic SMC proteins form two kind of heterodimers: the SMC1/SMC3 and the SMC2/SMC4 types. These heterodimers constitute an essential part of higher order complexes, which are involved in chromatin and DNA dynamics. This family also includes the RecF and RecN proteins that are involved in DNA metabolism and recombination.
Pssm-ID: 426784 [Multi-domain] Cd Length: 1161 Bit Score: 47.66 E-value: 4.01e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 186 ALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERK 265
Cdd:pfam02463 174 ALKKLIEETENLAELIIDLEELKLQELKLKEQAKKALEYYQLKEKLELEEEYLLYLDYLKLNEERIDLLQELLRDEQEEI 253
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 266 TMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKtdvEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRV 345
Cdd:pfam02463 254 ESSKQEIEKEEEKLAQVLKENKEEEKEKKLQEEELKLLAKEEEEL---KSELLKLERRKVDDEEKLKESEKEKKKAEKEL 330
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 346 NMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIKElsqevERLKLKLKDMKAIEDDLMKTEDEYETLERRYAN 425
Cdd:pfam02463 331 KKEKEEIEELEKELKELEIKREAEEEEEEELEKLQEKLEQLE-----EELLAKKKLESERLSSAAKLKEEELELKSEEEK 405
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 426 ERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKR--------LQEEEAKSGHLSREVDALKEKIHEYMATEDLIC 497
Cdd:pfam02463 406 EAQLLLELARQLEDLLKEEKKEELEILEEEEESIELKQGklteekeeLEKQELKLLKDELELKKSEDLLKETQLVKLQEQ 485
|
330 340 350 360
....*....|....*....|....*....|....*....|....*
gi 544346182 498 HLQGDHSVLQKKLNQQENRNRDLGREIENLTKELERYRHFSKSLR 542
Cdd:pfam02463 486 LELLLSRQKLEERSQKESKARSGLKVLLALIKDGVGGRIISAHGR 530
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
216-472 |
4.36e-05 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 46.68 E-value: 4.36e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 216 KQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLktltvmfvderktmSEKLKKTEDKLQAASSQLQVEQNKVTTV 295
Cdd:COG4942 23 AEAEAELEQLQQEIAELEKELAALKKEEKALLKQLAAL--------------ERRIAALARRIRALEQELAALEAELAEL 88
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 296 TEKlIEETKRALKsktdveekmysvtKERDDLKNKLKAEEEKGNdlLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKST 375
Cdd:COG4942 89 EKE-IAELRAELE-------------AQKEELAELLRALYRLGR--QPPLALLLSPEDFLDAVRRLQYLKYLAPARREQA 152
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 376 TALHQENNKIKELSQEVERLKLKLKDMKAieddlmKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTET 455
Cdd:COG4942 153 EELRADLAELAALRAELEAERAELEALLA------ELEEERAALEALKAERQKLLARLEKELAELAAELAELQQEAEELE 226
|
250
....*....|....*..
gi 544346182 456 SheqwLFKRLQEEEAKS 472
Cdd:COG4942 227 A----LIARLEAEAAAA 239
|
|
| Myosin_tail_1 |
pfam01576 |
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and ... |
16-541 |
4.45e-05 |
|
Myosin tail; The myosin molecule is a multi-subunit complex made up of two heavy chains and four light chains it is a fundamental contractile protein found in all eukaryote cell types. This family consists of the coiled-coil myosin heavy chain tail region. The coiled-coil is composed of the tail from two molecules of myosin. These can then assemble into the macromolecular thick filament. The coiled-coil region provides the structural backbone the thick filament.
Pssm-ID: 460256 [Multi-domain] Cd Length: 1081 Bit Score: 47.48 E-value: 4.45e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 16 QRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQID 95
Cdd:pfam01576 413 QLQELQARLSESERQRAELAEKLSKLQSELESVSSLLNEAEGKNIKLSKDVSSLESQLQDTQELLQEETRQKLNLSTRLR 492
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 96 ELEETNRSLRKAEEELQDIKEKISKgeygnagimaEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLEREtLQSKDFKL 175
Cdd:pfam01576 493 QLEDERNSLQEQLEEEEEAKRNVER----------QLSTLQAQLSDMKKKLEEDAGTLEALEEGKKRLQRE-LEALTQQL 561
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 176 EveklsKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRL----EKTEFTLKEDLT 251
Cdd:pfam01576 562 E-----EKAAAYDKLEKTKNRLQQELDDLLVDLDHQRQLVSNLEKKQKKFDQMLAEEKAISARYaeerDRAEAEAREKET 636
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 252 KLKTLTVMfVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLiEETKRALksktdvEEKMYSVTKERDDLKNKL 331
Cdd:pfam01576 637 RALSLARA-LEEALEAKEELERTNKQLRAEMEDLVSSKDDVGKNVHEL-ERSKRAL------EQQVEEMKTQLEELEDEL 708
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 332 KAEEEKgnDLLSRVNMlknrlQSLEAIEKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMK 411
Cdd:pfam01576 709 QATEDA--KLRLEVNM-----QALKAQFERDLQARDEQGEEKRRQLVKQVRELEAELEDERKQRAQAVAAKKKLELDLKE 781
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 412 TEDEYETLERRYAN-----ERDKAQF--LSKELEHVKME----LAKYKLAEKTETSHEQWLFkRLQEEEAKSGHLSREVD 480
Cdd:pfam01576 782 LEAQIDAANKGREEavkqlKKLQAQMkdLQRELEEARASrdeiLAQSKESEKKLKNLEAELL-QLQEDLAASERARRQAQ 860
|
490 500 510 520 530 540 550
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 481 ALKEKIHEYMAT---------------EDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKELERYRHFSKSL 541
Cdd:pfam01576 861 QERDELADEIASgasgksalqdekrrlEARIAQLEEELEEEQSNTELLNDRLRKSTLQVEQLTTELAAERSTSQKS 936
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
21-534 |
5.38e-05 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 46.98 E-value: 5.38e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 21 QELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQdqdtIMAKLTNEDSQNRQLQQKLAALSRQideleet 100
Cdd:TIGR02169 416 QRLSEELADLNAAIAGIEAKINELEEEKEDKALEIKKQEWKLEQ----LAADLSKYEQELYDLKEEYDRVEKE------- 484
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 101 nrsLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQ------------------------- 155
Cdd:TIGR02169 485 ---LSKLQRELAEAEAQARASEERVRGGRAVEEVLKASIQGVHGTVAQLGSVGERyataievaagnrlnnvvveddavak 561
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 156 -CRDLNKR--LERETL----QSKDFKLEVEKLSKR---------IMALEKLEDAFNKSKQECY------SLKCNLEKERM 213
Cdd:TIGR02169 562 eAIELLKRrkAGRATFlplnKMRDERRDLSILSEDgvigfavdlVEFDPKYEPAFKYVFGDTLvvedieAARRLMGKYRM 641
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 214 TT--------------------------KQLSQELESLKVRIKELEAIESRlekteftLKEDLTKLKTLTVMFVDERKTM 267
Cdd:TIGR02169 642 VTlegelfeksgamtggsraprggilfsRSEPAELQRLRERLEGLKRELSS-------LQSELRRIENRLDELSQELSDA 714
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 268 SEKLKKTEDKLQAASSQLQVEQNKVTTVTEKlIEETKRALkskTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNM 347
Cdd:TIGR02169 715 SRKIGEIEKEIEQLEQEEEKLKERLEELEED-LSSLEQEI---ENVKSELKELEARIEELEEDLHKLEEALNDLEARLSH 790
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 348 --LKNRLQSLEAIEKDFLK-----NKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLE 420
Cdd:TIGR02169 791 srIPEIQAELSKLEEEVSRiearlREIEQKLNRLTLEKEYLEKEIQELQEQRIDLKEQIKSIEKEIENLNGKKEELEEEL 870
|
490 500 510 520 530 540 550 560
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 421 RRYANE----RDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKEKIHE--YMATED 494
Cdd:TIGR02169 871 EELEAAlrdlESRLGDLKKERDELEAQLRELERKIEELEAQIEKKRKRLSELKAKLEALEEELSEIEDPKGEdeEIPEEE 950
|
570 580 590 600
....*....|....*....|....*....|....*....|....
gi 544346182 495 LichlqgDHSVLQKKLNQQENRNRDLG----REIENLTKELERY 534
Cdd:TIGR02169 951 L------SLEDVQAELQRVEEEIRALEpvnmLAIQEYEEVLKRL 988
|
|
| PRK12704 |
PRK12704 |
phosphodiesterase; Provisional |
104-253 |
7.94e-05 |
|
phosphodiesterase; Provisional
Pssm-ID: 237177 [Multi-domain] Cd Length: 520 Bit Score: 46.31 E-value: 7.94e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 104 LRKAEEELQDIKEKISKgeygnagimaEVEELRKRVLdMEGKDEELIKMEEQCRDLNKRleRETLQSKDFKLE--VEKLS 181
Cdd:PRK12704 33 IKEAEEEAKRILEEAKK----------EAEAIKKEAL-LEAKEEIHKLRNEFEKELRER--RNELQKLEKRLLqkEENLD 99
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 182 KRIMALEKLEDAFNKSKQECYSLKCNLEK-----ERMTTKQLsQELEslkvRIKEL---EAIESRLEKTEFTLKEDLTKL 253
Cdd:PRK12704 100 RKLELLEKREEELEKKEKELEQKQQELEKkeeelEELIEEQL-QELE----RISGLtaeEAKEILLEKVEEEARHEAAVL 174
|
|
| COG5283 |
COG5283 |
Phage-related tail protein [Mobilome: prophages, transposons]; |
7-163 |
7.99e-05 |
|
Phage-related tail protein [Mobilome: prophages, transposons];
Pssm-ID: 444094 [Multi-domain] Cd Length: 747 Bit Score: 46.39 E-value: 7.99e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 7 QRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMA---KLTNEDSQNRQL 83
Cdd:COG5283 10 KPFKSALESAKQRVAALAQALKALEAPTRALARALERAKQAAARLQTKYNKLRQSLQRLRQALDQagiDTRQLSAAQRRL 89
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 84 QQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISK-GEYGNAGI-------------------MAEVeelrKRVLDME 143
Cdd:COG5283 90 RSSLEQTNRQLERQQQRLARLGARQDRLKAARARLQRlAGAGAAAAaigaalaasvkpaidfedaMADV----AATVDLD 165
|
170 180
....*....|....*....|
gi 544346182 144 GKDEELIKMEEQCRDLNKRL 163
Cdd:COG5283 166 KSSEQFKALGKQARELSAQT 185
|
|
| COG5022 |
COG5022 |
Myosin heavy chain [General function prediction only]; |
26-515 |
9.38e-05 |
|
Myosin heavy chain [General function prediction only];
Pssm-ID: 227355 [Multi-domain] Cd Length: 1463 Bit Score: 46.61 E-value: 9.38e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 26 NAKETHTKLALAEARVQEE---EQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQ----------QKLAALSR 92
Cdd:COG5022 927 TELIARLKKLLNNIDLEEGpsiEYVKLPELNKLHEVESKLKETSEEYEDLLKKSTILVREGNkanselknfkKELAELSK 1006
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 93 QIDELEETNRSLRKAEEELQDIK--EKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETLQ- 169
Cdd:COG5022 1007 QYGALQESTKQLKELPVEVAELQsaSKIISSESTELSILKPLQKLKGLLLLENNQLQARYKALKLRRENSLLDDKQLYQl 1086
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 170 ------SKDFKLEVEKLSKRIMALEKLEDAFNKSKQecysLKCNLEKERMttkqlsqelESLKVRIKELEAIESRLEKTE 243
Cdd:COG5022 1087 estenlLKTINVKDLEVTNRNLVKPANVLQFIVAQM----IKLNLLQEIS---------KFLSQLVNTLEPVFQKLSVLQ 1153
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 244 FTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEqnkVTTVTEKLIEETKR-------------ALKSK 310
Cdd:COG5022 1154 LELDGLFWEANLEALPSPPPFAALSEKRLYQSALYDEKSKLSSSE---VNDLKNELIALFSKifsgwprgdklkkLISEG 1230
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 311 TDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQS-------LEAIEKDFLKN-----------KLNQDSG 372
Cdd:COG5022 1231 WVPTEYSTSLKGFNNLNKKFDTPASMSNEKLLSLLNSIDNLLSSykleeevLPATINSLLQYinvglfnalrtKASSLRW 1310
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 373 KSTTALHQ--ENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYanerdKAQFLSKELEHVKMeLAKYKLA 450
Cdd:COG5022 1311 KSATEVNYnsEELDDWCREFEISDVDEELEELIQAVKVLQLLKDDLNKLDELL-----DACYSLNPAEIQNL-KSRYDPA 1384
|
490 500 510 520 530 540
....*....|....*....|....*....|....*....|....*....|....*....|....*..
gi 544346182 451 EKtETSHEQWLFKRLQEEEAKS--GHLSREVDALKEKIHEYMATEDLICHLQGDHSVLQKKLNQQEN 515
Cdd:COG5022 1385 DK-ENNLPKEILKKIEALLIKQelQLSLEGKDETEVHLSEIFSEEKSLISLDRNSIYKEEVLSSLSA 1450
|
|
| PRK06975 |
PRK06975 |
bifunctional uroporphyrinogen-III synthetase/uroporphyrin-III C-methyltransferase; Reviewed |
6-112 |
9.80e-05 |
|
bifunctional uroporphyrinogen-III synthetase/uroporphyrin-III C-methyltransferase; Reviewed
Pssm-ID: 235899 [Multi-domain] Cd Length: 656 Bit Score: 46.25 E-value: 9.80e-05
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 6 QQRL--TAQLTLQRQkiqelttNAKETHTklalAEARVQEEEqkatrlekeLQTQTTKFHQDQDTIMAKLTNEDSQNRQL 83
Cdd:PRK06975 345 NRKVdrLDQELVQRQ-------QANDAQT----AELRVKTEQ---------AQASVHQLDSQFAQLDGKLADAQSAQQAL 404
|
90 100 110
....*....|....*....|....*....|.
gi 544346182 84 QQKLAALSRQID--ELEETNRSLRKAEEELQ 112
Cdd:PRK06975 405 EQQYQDLSRNRDdwMIAEVEQMLSSASQQLQ 435
|
|
| PRK12704 |
PRK12704 |
phosphodiesterase; Provisional |
16-187 |
1.03e-04 |
|
phosphodiesterase; Provisional
Pssm-ID: 237177 [Multi-domain] Cd Length: 520 Bit Score: 45.92 E-value: 1.03e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 16 QRQKIQELTTNAKETHTKLALAEARvQEEEQKATRLEKE-------LQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLA 88
Cdd:PRK12704 39 EAKRILEEAKKEAEAIKKEALLEAK-EEIHKLRNEFEKElrerrneLQKLEKRLLQKEENLDRKLELLEKREEELEKKEK 117
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 89 ALSRQIDELEETNRSLRKAEEELQDIKEKISkgeygnaGIMAevEELRKRVLDmegkdeeliKMEEQCRD----LNKRLE 164
Cdd:PRK12704 118 ELEQKQQELEKKEEELEELIEEQLQELERIS-------GLTA--EEAKEILLE---------KVEEEARHeaavLIKEIE 179
|
170 180
....*....|....*....|...
gi 544346182 165 RETlqskdfKLEVEKLSKRIMAL 187
Cdd:PRK12704 180 EEA------KEEADKKAKEILAQ 196
|
|
| rad50 |
TIGR00606 |
rad50; All proteins in this family for which functions are known are involvedin recombination, ... |
89-545 |
1.34e-04 |
|
rad50; All proteins in this family for which functions are known are involvedin recombination, recombinational repair, and/or non-homologous end joining.They are components of an exonuclease complex with MRE11 homologs. This family is distantly related to the SbcC family of bacterial proteins.This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University).
Pssm-ID: 129694 [Multi-domain] Cd Length: 1311 Bit Score: 45.81 E-value: 1.34e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 89 ALSRQIDELEETNRSLrKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETL 168
Cdd:TIGR00606 170 ALKQKFDEIFSATRYI-KALETLRQVRQTQGQKVQEHQMELKYLKQYKEKACEIRDQITSKEAQLESSREIVKSYENELD 248
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 169 QSKDFKLEVEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQEleslkvrikELEAIESRLEKTEFTLKE 248
Cdd:TIGR00606 249 PLKNRLKEIEHNLSKIMKLDNEIKALKSRKKQMEKDNSELELKMEKVFQGTDE---------QLNDLYHNHQRTVREKER 319
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 249 DLTKLKTLTVMFVDERKTMSEklKKTEDKLQAASSQLQVEQNKVTTVTEKLiEETKRALKSKTDVEEKMYSVTKErddLK 328
Cdd:TIGR00606 320 ELVDCQRELEKLNKERRLLNQ--EKTELLVEQGRLQLQADRHQEHIRARDS-LIQSLATRLELDGFERGPFSERQ---IK 393
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 329 NKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTT------ALHQENNKIKELSQEVERLKLKLKDM 402
Cdd:TIGR00606 394 NFHTLVIERQEDEAKTAAQLCADLQSKERLKQEQADEIRDEKKGLGRTielkkeILEKKQEELKFVIKELQQLEGSSDRI 473
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 403 KAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTE---------TSHEQWLFKRLQEEEAKSG 473
Cdd:TIGR00606 474 LELDQELRKAERELSKAEKNSLTETLKKEVKSLQNEKADLDRKLRKLDQEMEqlnhhtttrTQMEMLTKDKMDKDEQIRK 553
|
410 420 430 440 450 460 470
....*....|....*....|....*....|....*....|....*....|....*....|....*....|..
gi 544346182 474 HLSREVDALKEKIHEYMATEDLICHLQGdhsvLQKKLNQQENRNRDLGREIENLTKELERYRHFSKSLRPSL 545
Cdd:TIGR00606 554 IKSRHSDELTSLLGYFPNKKQLEDWLHS----KSKEINQTRDRLAKLNKELASLEQNKNHINNELESKEEQL 621
|
|
| COG2433 |
COG2433 |
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only]; |
88-256 |
1.86e-04 |
|
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only];
Pssm-ID: 441980 [Multi-domain] Cd Length: 644 Bit Score: 45.23 E-value: 1.86e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 88 AALSRQI-----DELEETNRSLRKAEEELQDIKEKISKGEygnagimAEVEELRKRVLDMEG----KDEELIKMEEQCRD 158
Cdd:COG2433 380 EALEELIekelpEEEPEAEREKEHEERELTEEEEEIRRLE-------EQVERLEAEVEELEAeleeKDERIERLERELSE 452
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 159 LNKRLERETLQSKdfklEVEKLSKRIMALEKledafnkskqecyslkcnlekermttkqlsqELESLKVRIKELEAIESR 238
Cdd:COG2433 453 ARSEERREIRKDR----EISRLDREIERLER-------------------------------ELEEERERIEELKRKLER 497
|
170
....*....|....*....
gi 544346182 239 LEKTEFTL-KEDLTKLKTL 256
Cdd:COG2433 498 LKELWKLEhSGELVPVKVV 516
|
|
| sbcc |
TIGR00618 |
exonuclease SbcC; All proteins in this family for which functions are known are part of an ... |
13-503 |
2.30e-04 |
|
exonuclease SbcC; All proteins in this family for which functions are known are part of an exonuclease complex with sbcD homologs. This complex is involved in the initiation of recombination to regulate the levels of palindromic sequences in DNA. This family is based on the phylogenomic analysis of JA Eisen (1999, Ph.D. Thesis, Stanford University). [DNA metabolism, DNA replication, recombination, and repair]
Pssm-ID: 129705 [Multi-domain] Cd Length: 1042 Bit Score: 44.96 E-value: 2.30e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 13 LTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQttkfHQDQDTIMAKLTNEDSQN--RQLQQKLAAL 90
Cdd:TIGR00618 403 DILQREQATIDTRTSAFRDLQGQLAHAKKQQELQQRYAELCAAAIT----CTAQCEKLEKIHLQESAQslKEREQQLQTK 478
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 91 SRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETLQS 170
Cdd:TIGR00618 479 EQIHLQETRKKAVVLARLLELQEEPCPLCGSCIHPNPARQDIDNPGPLTRRMQRGEQTYAQLETSEEDVYHQLTSERKQR 558
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 171 KDFKLEVEKLSKRIMALEKLEDAF----NKSKQECYSLKCNLEKERMTTKQLSQELESLKVRiKELEAIESRLEKTEFTL 246
Cdd:TIGR00618 559 ASLKEQMQEIQQSFSILTQCDNRSkediPNLQNITVRLQDLTEKLSEAEDMLACEQHALLRK-LQPEQDLQDVRLHLQQC 637
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 247 KEDLTKLKT------LTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLiEETKRALKSKTDVEEKMYSV 320
Cdd:TIGR00618 638 SQELALKLTalhalqLTLTQERVREHALSIRVLPKELLASRQLALQKMQSEKEQLTYWK-EMLAQCQTLLRELETHIEEY 716
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 321 TKERDDLKNKLKAeeeKGNDLLSRVNMLKNRLQSLEAIEKDFLKNKLNQDSGKSTTALHQE-------------NNKIKE 387
Cdd:TIGR00618 717 DREFNEIENASSS---LGSDLAAREDALNQSLKELMHQARTVLKARTEAHFNNNEEVTAALqtgaelshlaaeiQFFNRL 793
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 388 LSQEVERLKLKLKDMKA-IEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEQWLFKRLQ 466
Cdd:TIGR00618 794 REEDTHLLKTLEAEIGQeIPSDEDILNLQCETLVQEEEQFLSRLEEKSATLGEITHQLLKYEECSKQLAQLTQEQAKIIQ 873
|
490 500 510
....*....|....*....|....*....|....*....
gi 544346182 467 EEEAKSGHLSREVDALKEKIHEYMA--TEDLICHLQGDH 503
Cdd:TIGR00618 874 LSDKLNGINQIKIQFDGDALIKFLHeiTLYANVRLANQS 912
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
9-166 |
2.40e-04 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 45.01 E-value: 2.40e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 9 LTAQLTLQRQKIQELTTnakethtKLALAEARVQEEEQKATRLEKELqtqttkfhqDQDTIMAKLTNEDSQNRQLQQKLA 88
Cdd:COG3206 210 LSEEAKLLLQQLSELES-------QLAEARAELAEAEARLAALRAQL---------GSGPDALPELLQSPVIQQLRAQLA 273
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 89 ALSRQIDELE----ETNRSLRKAEEELQDIKEKISK-GEYGNAGIMAEVEELRKRVLDMEgkdEELIKMEEQCRDLNK-- 161
Cdd:COG3206 274 ELEAELAELSarytPNHPDVIALRAQIAALRAQLQQeAQRILASLEAELEALQAREASLQ---AQLAQLEARLAELPEle 350
|
....*....
gi 544346182 162 ----RLERE 166
Cdd:COG3206 351 aelrRLERE 359
|
|
| Smc |
COG1196 |
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning]; ... |
5-169 |
2.52e-04 |
|
Chromosome segregation ATPase Smc [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 440809 [Multi-domain] Cd Length: 983 Bit Score: 44.93 E-value: 2.52e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 5 EQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKfHQDQDTIMAKLTNEDSQNRQLQ 84
Cdd:COG1196 664 GGSRRELLAALLEAEAELEELAERLAEEELELEEALLAEEEEERELAEAEEERLEEE-LEEEALEEQLEAEREELLEELL 742
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 85 QKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLE 164
Cdd:COG1196 743 EEEELLEEEALEELPEPPDLEELERELERLEREIEALGPVNLLAIEEYEELEERYDFLSEQREDLEEARETLEEAIEEID 822
|
....*
gi 544346182 165 RETLQ 169
Cdd:COG1196 823 RETRE 827
|
|
| CCCAP |
pfam15964 |
Centrosomal colon cancer autoantigen protein family; CCCAP is a family of proteins found in ... |
4-367 |
3.48e-04 |
|
Centrosomal colon cancer autoantigen protein family; CCCAP is a family of proteins found in eukaryotes. CCCAP is also known as SDCCAG8, serologically defined colon cancer antigen 8. It is associated with the centrosome.
Pssm-ID: 435040 [Multi-domain] Cd Length: 703 Bit Score: 44.51 E-value: 3.48e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQ- 82
Cdd:pfam15964 325 EAQQRESSAYEQVKQAVQMTEEANFEKTKALIQCEQLKSELERQKERLEKELASQQEKRAQEKEALRKEMKKEREELGAt 404
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 83 ---LQQKLAALSRQIDELEETNRSLRKAEEELQ---------------DIKEKISKGEYGNAGIMAEVEELRKRVL-DME 143
Cdd:pfam15964 405 mlaLSQNVAQLEAQVEKVTREKNSLVSQLEEAQkqlasqemdvtkvcgEMRYQLNQTKMKKDEAEKEHREYRTKTGrQLE 484
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 144 GKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKrimALEKLEDAFNKSKQECYSLKCNLEKE-RMTTKQLSQEL 222
Cdd:pfam15964 485 IKDQEIEKLGLELSESKQRLEQAQQDAARAREECLKLTE---LLGESEHQLHLTRLEKESIQQSFSNEaKAQALQAQQRE 561
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 223 ESLKVRIKELEAiesRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEE 302
Cdd:pfam15964 562 QELTQKMQQMEA---QHDKTVNEQYSLLTSQNTFIAKLKEECCTLAKKLEEITQKSRSEVEQLSQEKEYLQDRLEKLQKR 638
|
330 340 350 360 370 380
....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 303 TKRaLKSKTDVEEKMYSVTKER-DDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLeAIEKDFLKNKL 367
Cdd:pfam15964 639 NEE-LEEQCVQHGRMHERMKQRlRQLDKHCQATAQQLVQLLSKQNQLFKERQNL-TEEVQSLRSQV 702
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
4-169 |
4.00e-04 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 44.24 E-value: 4.00e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLE-----KELQTQTTKFHQDQDTIMAKLTNEDS 78
Cdd:COG3206 212 EEAKLLLQQLSELESQLAEARAELAEAEARLAALRAQLGSGPDALPELLqspviQQLRAQLAELEAELAELSARYTPNHP 291
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 79 QNRQLQQKLAALSRQIDelEETNRSLRKAEEELQDIKEKIskgeygnAGIMAEVEELRKRVLDMEGKDEELIKMEEQcRD 158
Cdd:COG3206 292 DVIALRAQIAALRAQLQ--QEAQRILASLEAELEALQARE-------ASLQAQLAQLEARLAELPELEAELRRLERE-VE 361
|
170
....*....|.
gi 544346182 159 LNKRLERETLQ 169
Cdd:COG3206 362 VARELYESLLQ 372
|
|
| GumC |
COG3206 |
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis]; |
75-297 |
4.47e-04 |
|
Exopolysaccharide export protein/domain GumC/Wzc1 [Cell wall/membrane/envelope biogenesis];
Pssm-ID: 442439 [Multi-domain] Cd Length: 687 Bit Score: 43.85 E-value: 4.47e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 75 NEDSQNRQLQQKLAALSRQIDELEEtnrSLRKAEEELQDIKEK---ISKGEYGNAgIMAEVEELRKRVLDMEgkdEELIK 151
Cdd:COG3206 165 NLELRREEARKALEFLEEQLPELRK---ELEEAEAALEEFRQKnglVDLSEEAKL-LLQQLSELESQLAEAR---AELAE 237
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 152 MEEQCRDLNKRLERETLQSKDFK--LEVEKLSKRIMALEKLEDAFNKS-----------KQECYSLKCNLEKE-RMTTKQ 217
Cdd:COG3206 238 AEARLAALRAQLGSGPDALPELLqsPVIQQLRAQLAELEAELAELSARytpnhpdvialRAQIAALRAQLQQEaQRILAS 317
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 218 LSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTvmfvDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTE 297
Cdd:COG3206 318 LEAELEALQAREASLQAQLAQLEARLAELPELEAELRRLE----REVEVARELYESLLQRLEEARLAEALTVGNVRVIDP 393
|
|
| COG2433 |
COG2433 |
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only]; |
295-482 |
5.06e-04 |
|
Possible nuclease of RNase H fold, RuvC/YqgF family [General function prediction only];
Pssm-ID: 441980 [Multi-domain] Cd Length: 644 Bit Score: 43.69 E-value: 5.06e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 295 VTEKLIEEtkralksKTDVEEKMYSVTKERDDlkNKLKAEEEKGNDLLSRVNMLKNRLQSLEaiekdflknklnqdsgks 374
Cdd:COG2433 381 ALEELIEK-------ELPEEEPEAEREKEHEE--RELTEEEEEIRRLEEQVERLEAEVEELE------------------ 433
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 375 ttalhqenNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANerdkaqfLSKELEHVKMELAkyKLAEKTE 454
Cdd:COG2433 434 --------AELEEKDERIERLERELSEARSEERREIRKDREISRLDREIER-------LERELEEERERIE--ELKRKLE 496
|
170 180
....*....|....*....|....*....
gi 544346182 455 tsheqwLFKRLQEEEAKSGHLS-REVDAL 482
Cdd:COG2433 497 ------RLKELWKLEHSGELVPvKVVEKF 519
|
|
| YhaN |
COG4717 |
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown]; |
3-200 |
5.10e-04 |
|
Uncharacterized conserved protein YhaN, contains AAA domain [Function unknown];
Pssm-ID: 443752 [Multi-domain] Cd Length: 641 Bit Score: 43.60 E-value: 5.10e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 3 VDEQQRLTAQLTLQRQKIQEL-------TTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTN 75
Cdd:COG4717 304 AEELQALPALEELEEEELEELlaalglpPDLSPEELLELLDRIEELQELLREAEELEEELQLEELEQEIAALLAEAGVED 383
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 76 EDS---------QNRQLQQKLAALSRQIDELEETNRSLRKA------EEELQDIKEKISKGEygnagimAEVEELRKRVL 140
Cdd:COG4717 384 EEElraaleqaeEYQELKEELEELEEQLEELLGELEELLEAldeeelEEELEELEEELEELE-------EELEELREELA 456
|
170 180 190 200 210 220
....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 141 DMEgkdEELIKMEEQCRDLNKRLERETLQSKdFKLEVEKLSKRIMALEKLEDAFNKSKQE 200
Cdd:COG4717 457 ELE---AELEQLEEDGELAELLQELEELKAE-LRELAEEWAALKLALELLEEAREEYREE 512
|
|
| PRK01156 |
PRK01156 |
chromosome segregation protein; Provisional |
67-576 |
5.90e-04 |
|
chromosome segregation protein; Provisional
Pssm-ID: 100796 [Multi-domain] Cd Length: 895 Bit Score: 43.74 E-value: 5.90e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 67 DTIMAKLTNEDSQNRQLQQK---LAALSRQIDELEETNRSLRKAEEELQDikekiskgEYGNAgiMAEVEELRKRVLDME 143
Cdd:PRK01156 176 DMLRAEISNIDYLEEKLKSSnleLENIKKQIADDEKSHSITLKEIERLSI--------EYNNA--MDDYNNLKSALNELS 245
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 144 GKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRImaleklEDAFNKSKQEC---YSLKCNLEKERMTTKQLSQ 220
Cdd:PRK01156 246 SLEDMKNRYESEIKTAESDLSMELEKNNYYKELEERHMKII------NDPVYKNRNYIndyFKYKNDIENKKQILSNIDA 319
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 221 ELESLKVRIKELEAIESRLEKTEftlkedltklktltvmfvdERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLI 300
Cdd:PRK01156 320 EINKYHAIIKKLSVLQKDYNDYI-------------------KKKSRYDDLNNQILELEGYEMDYNSYLKSIESLKKKIE 380
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 301 EETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAiEKDFLKNKLNQDSGKST----- 375
Cdd:PRK01156 381 EYSKNIERMSAFISEILKIQEIDPDAIKKELNEINVKLQDISSKVSSLNQRIRALRE-NLDELSRNMEMLNGQSVcpvcg 459
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 376 TALHQE---------NNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLE-RRYANERDKAQFLSKELEHVKMELA 445
Cdd:PRK01156 460 TTLGEEksnhiinhyNEKKSRLEEKIREIEIEVKDIDEKIVDLKKRKEYLESEEiNKSINEYNKIESARADLEDIKIKIN 539
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 446 KYKlaektetsheqwlFKRLQEEEAKSGHLSREVDALKEKIHEY---MATEDLIC--HLQGDHSVLQKKLNQQENRNRDL 520
Cdd:PRK01156 540 ELK-------------DKHDKYEEIKNRYKSLKLEDLDSKRTSWlnaLAVISLIDieTNRSRSNEIKKQLNDLESRLQEI 606
|
490 500 510 520 530
....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 521 GREIENLTKELERYrhfSKSLRPSLNGRRISDPQVFSKEVQTEAVDNEPPDYKSLI 576
Cdd:PRK01156 607 EIGFPDDKSYIDKS---IREIENEANNLNNKYNEIQENKILIEKLRGKIDNYKKQI 659
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
2-103 |
6.25e-04 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 43.21 E-value: 6.25e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 2 VVDEQQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNR 81
Cdd:COG4942 144 LAPARREQAEELRADLAELAALRAELEAERAELEALLAELEEERAALEALKAERQKLLARLEKELAELAAELAELQQEAE 223
|
90 100
....*....|....*....|..
gi 544346182 82 QLQQKLAALSRQIDELEETNRS 103
Cdd:COG4942 224 ELEALIARLEAEAAAAAERTPA 245
|
|
| DR0291 |
COG1579 |
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General ... |
110-310 |
7.87e-04 |
|
Predicted nucleic acid-binding protein DR0291, contains C4-type Zn-ribbon domain [General function prediction only];
Pssm-ID: 441187 [Multi-domain] Cd Length: 236 Bit Score: 42.22 E-value: 7.87e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 110 ELQDIKEKISKgeygnagIMAEVEELRKRVLDMEgkdEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEK 189
Cdd:COG1579 11 DLQELDSELDR-------LEHRLKELPAELAELE---DELAALEARLEAAKTELEDLEKEIKRLELEIEEVEARIKKYEE 80
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 190 LEDAFNKSKQecYslkcnlekermttKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKtltvmfvDERKTMSE 269
Cdd:COG1579 81 QLGNVRNNKE--Y-------------EALQKEIESLKRRISDLEDEILELMERIEELEEELAELE-------AELAELEA 138
|
170 180 190 200
....*....|....*....|....*....|....*....|.
gi 544346182 270 KLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSK 310
Cdd:COG1579 139 ELEEKKAELDEELAELEAELEELEAEREELAAKIPPELLAL 179
|
|
| PRK05771 |
PRK05771 |
V-type ATP synthase subunit I; Validated |
267-486 |
8.71e-04 |
|
V-type ATP synthase subunit I; Validated
Pssm-ID: 235600 [Multi-domain] Cd Length: 646 Bit Score: 42.99 E-value: 8.71e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 267 MSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERddlKNKLKAEEEKGNDLLSRVN 346
Cdd:PRK05771 51 LLTKLSEALDKLRSYLPKLNPLREEKKKVSVKSLEELIKDVEEELEKIEKEIKELEEE---ISELENEIKELEQEIERLE 127
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 347 MLKN------RLQSLE-------AIEKDFLKNKLNQDSGKSTTALHQENNKI-------KELSQEVERLkLKLKDMKAIE 406
Cdd:PRK05771 128 PWGNfdldlsLLLGFKyvsvfvgTVPEDKLEELKLESDVENVEYISTDKGYVyvvvvvlKELSDEVEEE-LKKLGFERLE 206
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 407 -DDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAEKTETSHEqwlfkrLQEEE-----AKSGHL----- 475
Cdd:PRK05771 207 lEEEGTPSELIREIKEELEEIEKERESLLEELKELAKKYLEELLALYEYLEIE------LERAEalskfLKTDKTfaieg 280
|
250
....*....|....
gi 544346182 476 ---SREVDALKEKI 486
Cdd:PRK05771 281 wvpEDRVKKLKELI 294
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
6-189 |
8.86e-04 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 42.98 E-value: 8.86e-04
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 6 QQRLTAQLTLQRQKIQELTTNAKETHTKLALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTImakltnEDSQNRQLQ- 84
Cdd:COG4913 262 ERYAAARERLAELEYLRAALRLWFAQRRLELLEAELEELRAELARLEAELERLEARLDALREEL------DELEAQIRGn 335
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 85 --QKLAALSRQIDELEETnrsLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVldmegkDEELIKMEEQCRDLNKR 162
Cdd:COG4913 336 ggDRLEQLEREIERLERE---LEERERRRARLEALLAALGLPLPASAEEFAALRAEA------AALLEALEEELEALEEA 406
|
170 180
....*....|....*....|....*..
gi 544346182 163 LERETLQSKDFKLEVEKLSKRIMALEK 189
Cdd:COG4913 407 LAEAEAALRDLRRELRELEAEIASLER 433
|
|
| 235kDa-fam |
TIGR01612 |
reticulocyte binding/rhoptry protein; This model represents a group of paralogous families in ... |
70-460 |
1.05e-03 |
|
reticulocyte binding/rhoptry protein; This model represents a group of paralogous families in plasmodium species alternately annotated as reticulocyte binding protein, 235-kDa family protein and rhoptry protein. Rhoptry protein is localized on the cell surface and is extremely large (although apparently lacking in repeat structure) and is important for the process of invasion of the RBCs by the parasite. These proteins are found in P. falciparum, P. vivax and P. yoelii.
Pssm-ID: 130673 [Multi-domain] Cd Length: 2757 Bit Score: 43.12 E-value: 1.05e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 70 MAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEE-LQDIKEKISKGEYGN--AGIMAE---VEELRKRVLD-- 141
Cdd:TIGR01612 485 IDENSKQDNTVKLILMRMKDFKDIIDFMELYKPDEVPSKNIiGFDIDQNIKAKLYKEieAGLKESyelAKNWKKLIHEik 564
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 142 --MEGKDEELIKMEEQCRDL-NKRLE--RETLQSKDFKLEVEklskrimalEKLEDAFNKSK--QECYSLKCNLEKERMT 214
Cdd:TIGR01612 565 keLEEENEDSIHLEKEIKDLfDKYLEidDEIIYINKLKLELK---------EKIKNISDKNEyiKKAIDLKKIIENNNAY 635
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 215 TKQLS-----QELESLKVRIKELEAIESRLEKTeftLKEDLTKLKTLTVMFVDErktmsEKLKKTEDK--LQAASSQLQV 287
Cdd:TIGR01612 636 IDELAkispyQVPEHLKNKDKIYSTIKSELSKI---YEDDIDALYNELSSIVKE-----NAIDNTEDKakLDDLKSKIDK 707
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 288 EQNKVTTVTEKLIE------ETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNrlqslEAIEKD 361
Cdd:TIGR01612 708 EYDKIQNMETATVElhlsniENKKNELLDIIVEIKKHIHGEINKDLNKILEDFKNKEKELSNKINDYAK-----EKDELN 782
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 362 FLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLerryanerdKAQFLSKELEHVK 441
Cdd:TIGR01612 783 KYKSKISEIKNHYNDQINIDNIKDEDAKQNYDKSKEYIKTISIKEDEIFKIINEMKFM---------KDDFLNKVDKFIN 853
|
410
....*....|....*....
gi 544346182 442 MElakYKLAEKTETSHEQW 460
Cdd:TIGR01612 854 FE---NNCKEKIDSEHEQF 869
|
|
| Spc7 |
smart00787 |
Spc7 kinetochore protein; This domain is found in cell division proteins which are required ... |
51-190 |
1.08e-03 |
|
Spc7 kinetochore protein; This domain is found in cell division proteins which are required for kinetochore-spindle association.
Pssm-ID: 197874 [Multi-domain] Cd Length: 312 Bit Score: 41.93 E-value: 1.08e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 51 LEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKA-EEELQDIKEKISKGEYGNAGIM 129
Cdd:smart00787 145 LKEGLDENLEGLKEDYKLLMKELELLNSIKPKLRDRKDALEEELRQLKQLEDELEDCdPTELDRAKEKLKKLLQEIMIKV 224
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|..
gi 544346182 130 AEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFKL-EVEKLSKRIMALEKL 190
Cdd:smart00787 225 KKLEELEEELQELESKIEDLTNKKSELNTEIAEAEKKLEQCRGFTFkEIEKLKEQLKLLQSL 286
|
|
| MukB |
COG3096 |
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell ... |
4-292 |
1.34e-03 |
|
Chromosome condensin MukBEF, ATPase and DNA-binding subunit MukB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 442330 [Multi-domain] Cd Length: 1470 Bit Score: 42.63 E-value: 1.34e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELttnaKETHTKLA--LAEARVQEEEQKATRLEkELQTQTTKFHQDQDTIMAKltneDSQNR 81
Cdd:COG3096 850 RELAQHRAQEQQLRQQLDQL----KEQLQLLNklLPQANLLADETLADRLE-ELREELDAAQEAQAFIQQH----GKALA 920
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 82 QLQQKLAALSRQIDELEETNRSLRKAEEELQDIKekiskgeygnAGIMAeVEELRKRVLDMEGKDEEliKMEEQCRDLNK 161
Cdd:COG3096 921 QLEPLVAVLQSDPEQFEQLQADYLQAKEQQRRLK----------QQIFA-LSEVVQRRPHFSYEDAV--GLLGENSDLNE 987
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 162 RLERETLQSKdfklevEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEK 241
Cdd:COG3096 988 KLRARLEQAE------EARREAREQLRQAQAQYSQYNQVLASLKSSRDAKQQTLQELEQELEELGVQADAEAEERARIRR 1061
|
250 260 270 280 290
....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 242 TEftLKEDLTKL--------KTLTVmFVDERKTMSEKLKKTEDKLQAASSqlQVEQNKV 292
Cdd:COG3096 1062 DE--LHEELSQNrsrrsqleKQLTR-CEAEMDSLQKRLRKAERDYKQERE--QVVQAKA 1115
|
|
| COG4913 |
COG4913 |
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown]; |
81-336 |
1.40e-03 |
|
Uncharacterized conserved protein, contains a C-terminal ATPase domain [Function unknown];
Pssm-ID: 443941 [Multi-domain] Cd Length: 1089 Bit Score: 42.59 E-value: 1.40e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 81 RQLQQKLAALSRQIDELEETNRSLRKAEEE---LQDIKEKisKGEYGNAGIMAEVEELRKRVLDMEGKDEELIKMEEQCR 157
Cdd:COG4913 221 PDTFEAADALVEHFDDLERAHEALEDAREQielLEPIREL--AERYAAARERLAELEYLRAALRLWFAQRRLELLEAELE 298
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 158 DLNKRLERETLQSKDFKLEVEKLSKRIMALEK-LEDAFNKSKQEcyslkcnLEKERmttKQLSQELESLKVRIKELEAIE 236
Cdd:COG4913 299 ELRAELARLEAELERLEARLDALREELDELEAqIRGNGGDRLEQ-------LEREI---ERLERELEERERRRARLEALL 368
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 237 SRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQnkvttvtEKLIEETKRALKSKTDVEEK 316
Cdd:COG4913 369 AALGLPLPASAEEFAALRAEAAALLEALEEELEALEEALAEAEAALRDLRREL-------RELEAEIASLERRKSNIPAR 441
|
250 260
....*....|....*....|
gi 544346182 317 MYSVtkeRDDLKNKLKAEEE 336
Cdd:COG4913 442 LLAL---RDALAEALGLDEA 458
|
|
| PTZ00121 |
PTZ00121 |
MAEBL; Provisional |
38-340 |
1.48e-03 |
|
MAEBL; Provisional
Pssm-ID: 173412 [Multi-domain] Cd Length: 2084 Bit Score: 42.44 E-value: 1.48e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 38 EARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQlqqKLAALSRQIDELEETNRSLRKAEEELQDIKEK 117
Cdd:PTZ00121 1617 EAKIKAEELKKAEEEKKKVEQLKKKEAEEKKKAEELKKAEEENKI---KAAEEAKKAEEDKKKAEEAKKAEEDEKKAAEA 1693
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 118 ISKGEYGNAgimaEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKrimalEKLEDAFNKS 197
Cdd:PTZ00121 1694 LKKEAEEAK----KAEELKKKEAEEKKKAEELKKAEEENKIKAEEAKKEAEEDKKKAEEAKKDEE-----EKKKIAHLKK 1764
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 198 KQECYSLKCNLEKERMTTKQLSQELESLKVRI-KELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMS-------- 268
Cdd:PTZ00121 1765 EEEKKAEEIRKEKEAVIEEELDEEDEKRRMEVdKKIKDIFDNFANIIEGGKEGNLVINDSKEMEDSAIKEVAdsknmqle 1844
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 269 -----EKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALK----SKTDVEEKMYSVT---KERDDLKNKLKAEEE 336
Cdd:PTZ00121 1845 eadafEKHKFNKNNENGEDGNKEADFNKEKDLKEDDEEEIEEADEiekiDKDDIEREIPNNNmagKNNDIIDDKLDKDEY 1924
|
....
gi 544346182 337 KGND 340
Cdd:PTZ00121 1925 IKRD 1928
|
|
| 46 |
PHA02562 |
endonuclease subunit; Provisional |
3-161 |
1.52e-03 |
|
endonuclease subunit; Provisional
Pssm-ID: 222878 [Multi-domain] Cd Length: 562 Bit Score: 42.31 E-value: 1.52e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 3 VDEQQRLTAQLTLQRQ-KIQELTTNAKETHTKLALAEARVQEEE--------------QKATRLEKELQTQT--TKFHQD 65
Cdd:PHA02562 204 IEEQRKKNGENIARKQnKYDELVEEAKTIKAEIEELTDELLNLVmdiedpsaalnklnTAAAKIKSKIEQFQkvIKMYEK 283
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 66 QDT-------------IMAKLTNedsQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYG-------N 125
Cdd:PHA02562 284 GGVcptctqqisegpdRITKIKD---KLKELQHSLEKLDTAIDELEEIMDEFNEQSKKLLELKNKISTNKQSlitlvdkA 360
|
170 180 190
....*....|....*....|....*....|....*.
gi 544346182 126 AGIMAEVEELRKRVLDmegKDEELIKMEEQCRDLNK 161
Cdd:PHA02562 361 KKVKAAIEELQAEFVD---NAEELAKLQDELDKIVK 393
|
|
| PTZ00440 |
PTZ00440 |
reticulocyte binding protein 2-like protein; Provisional |
82-530 |
1.56e-03 |
|
reticulocyte binding protein 2-like protein; Provisional
Pssm-ID: 240419 [Multi-domain] Cd Length: 2722 Bit Score: 42.51 E-value: 1.56e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 82 QLQQKLAALSRQIDELEEtnrSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRvLDMEGKDEELIKMEEQCRDLNK 161
Cdd:PTZ00440 788 TILNKENKISNDINILKE---NKKNNQDLLNSYNILIQKLEAHTEKNDEELKQLLQK-FPTEDENLNLKELEKEFNENNQ 863
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 162 RLErETLQskdfklEVEKLSKRIMALEKLEDAFNKS---KQECYSLKCNLEKERMTTKQLSQELES------------LK 226
Cdd:PTZ00440 864 IVD-NIIK------DIENMNKNINIIKTLNIAINRSnsnKQLVEHLLNNKIDLKNKLEQHMKIINTdniiqkneklnlLN 936
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 227 VRIKELEAIESRLEKTEFT-----LKEDLTKLKTLTVMFVDERKTMSEKLKKTED-------KLQAASSQLQVEQNKVTT 294
Cdd:PTZ00440 937 NLNKEKEKIEKQLSDTKINnlkmqIEKTLEYYDKSKENINGNDGTHLEKLDKEKDewehfksEIDKLNVNYNILNKKIDD 1016
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 295 VT----EKLIEETKRALKSK-TDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKDFLKNklnq 369
Cdd:PTZ00440 1017 LIkkqhDDIIELIDKLIKEKgKEIEEKVDQYISLLEKMKTKLSSFHFNIDIKKYKNPKIKEEIKLLEEKVEALLKK---- 1092
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 370 dsgksttalhqennkIKELSQEVERLKLKLKDMKAIEDDLM-KTEDEYET----LERRYANERDkaqfLSKELEHVKMEL 444
Cdd:PTZ00440 1093 ---------------IDENKNKLIEIKNKSHEHVVNADKEKnKQTEHYNKkkksLEKIYKQMEK----TLKELENMNLED 1153
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 445 AKYKLAEKTETSHEQWLF----KRLQEEEAKSGHLSREVDALKEKIHEYMAteDLICHLQGDHSVLqkKLNQQENRNRDL 520
Cdd:PTZ00440 1154 ITLNEVNEIEIEYERILIdhivEQINNEAKKSKTIMEEIESYKKDIDQVKK--NMSKERNDHLTTF--EYNAYYDKATAS 1229
|
490
....*....|
gi 544346182 521 GREIENLTKE 530
Cdd:PTZ00440 1230 YENIEELTTE 1239
|
|
| 46 |
PHA02562 |
endonuclease subunit; Provisional |
246-484 |
1.80e-03 |
|
endonuclease subunit; Provisional
Pssm-ID: 222878 [Multi-domain] Cd Length: 562 Bit Score: 41.92 E-value: 1.80e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 246 LKEDLTKLKTLTVMfvderktmsEKLKKteDKLQAASSQLQVEQNKVTTVTEKL------IEETKRalKSKTDVEEKMYS 319
Cdd:PHA02562 155 LVEDLLDISVLSEM---------DKLNK--DKIRELNQQIQTLDMKIDHIQQQIktynknIEEQRK--KNGENIARKQNK 221
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 320 VTKERDDLKNkLKAEEEKGNDLLSRVNM------------------LKNRLQSLEAIEKDFLKNklnQDSGKSTTALHQE 381
Cdd:PHA02562 222 YDELVEEAKT-IKAEIEELTDELLNLVMdiedpsaalnklntaaakIKSKIEQFQKVIKMYEKG---GVCPTCTQQISEG 297
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 382 NNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEY--------------ETLERRYANERDKAQFLSKELEHVKMELAKY 447
Cdd:PHA02562 298 PDRITKIKDKLKELQHSLEKLDTAIDELEEIMDEFneqskkllelknkiSTNKQSLITLVDKAKKVKAAIEELQAEFVDN 377
|
250 260 270
....*....|....*....|....*....|....*..
gi 544346182 448 KLAEKTETSHEQWLFKRLQEEEAKSGHLSREVDALKE 484
Cdd:PHA02562 378 AEELAKLQDELDKIVKTKSELVKEKYHRGIVTDLLKD 414
|
|
| CagA_N |
pfam18971 |
CagA protein; The Helicobacter pylori type IV secretion effector CagA is a major bacterial ... |
6-292 |
1.92e-03 |
|
CagA protein; The Helicobacter pylori type IV secretion effector CagA is a major bacterial virulence determinant and critical for gastric carcinogenesis. X-ray crystallographic analysis of the N-terminal CagA fragment (residues 1-876) revealed that the region has a structure comprised of three discrete domains. Domain I constitutes a mobile CagA N terminus, while Domain II tethers CagA to the plasma membrane by interacting with membrane phosphatidylserine. Domain III interacts intramolecularly with the intrinsically disordered C-terminal region, and this interaction potentiates the pathogenic scaffold/hub function of CagA.
Pssm-ID: 408741 [Multi-domain] Cd Length: 876 Bit Score: 42.07 E-value: 1.92e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 6 QQRLTAQ-LTLQRQK--IQELTTNAKETHTKL-----ALAEARVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNED 77
Cdd:pfam18971 562 ENKLTAKgLSLQEANklIKDFLSSNKELAGKAlnfnkAVAEAKSTGNYDEVKKAQKDLEKSLRKREHLEKEVEKKLESKS 641
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 78 SQNRQLQQKLAALSrQIDEL-----EETNR---------SLRKAEEELQDIKEKISKgeygnagimaEVEELRKRVLDME 143
Cdd:pfam18971 642 GNKNKMEAKAQANS-QKDEIfalinKEANRdaraiaytqNLKGIKRELSDKLEKISK----------DLKDFSKSFDEFK 710
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 144 -GKDEELIKMEEQCRDLNKRLeretlqsKDFKLEVEKLSK---RIMALEKLEDAFNKSKQECYSLKCNLE---KERMTTK 216
Cdd:pfam18971 711 nGKNKDFSKAEETLKALKGSV-------KDLGINPEWISKvenLNAALNEFKNGKNKDFSKVTQAKSDLEnsvKDVIINQ 783
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 217 QLSQELESLKVRIKELEAIE--SRLEKTeftlkedLTKLKTLTvmfvdeRKTMSEKLKKTEDKLQAASSQL-QVEQNKV 292
Cdd:pfam18971 784 KVTDKVDNLNQAVSVAKAMGdfSRVEQV-------LADLKNFS------KEQLAQQAQKNEDFNTGKNSELyQSVKNSV 849
|
|
| DUF4407 |
pfam14362 |
Domain of unknown function (DUF4407); This family of proteins is found in bacteria. Proteins ... |
52-193 |
2.15e-03 |
|
Domain of unknown function (DUF4407); This family of proteins is found in bacteria. Proteins in this family are typically between 366 and 597 amino acids in length. There is a single completely conserved residue R that may be functionally important.
Pssm-ID: 464151 [Multi-domain] Cd Length: 295 Bit Score: 41.08 E-value: 2.15e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 52 EKELQTQ-TTKFHQDQDTIMAKLTNE-DSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKGEYGNAGI- 128
Cdd:pfam14362 105 EKEIDRElLEIQQEEADAAKAQLAAAyRARLAELEAQIAALDAEIDAAEARLDALQAEARCELDGTPGTGTGVPGDGPVa 184
|
90 100 110 120 130 140
....*....|....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 129 ---MAEVEELRKRVLDMEGK-DEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDA 193
Cdd:pfam14362 185 ktkQAQLDAAQAELAALQAQnDARLAALRAELARLTAERAAARARSQAAIDGDDGLLARLEALNRLTTE 253
|
|
| PRK10246 |
PRK10246 |
exonuclease subunit SbcC; Provisional |
4-240 |
2.86e-03 |
|
exonuclease subunit SbcC; Provisional
Pssm-ID: 182330 [Multi-domain] Cd Length: 1047 Bit Score: 41.32 E-value: 2.86e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 4 DEQQRLTAQLTLQRQKIQELTTNAKEThtkLALAEARVQEEEQKATRLEKELQTQTTKFHQDQD------------TIMA 71
Cdd:PRK10246 297 ERIQEQSAALAHTRQQIEEVNTRLQST---MALRARIRHHAAKQSAELQAQQQSLNTWLAEHDRfrqwnnelagwrAQFS 373
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 72 KLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKaeeelQDIKEKISKgeygnagiMAEVEELRKRvldmegkdeeLIK 151
Cdd:PRK10246 374 QQTSDREQLRQWQQQLTHAEQKLNALPAITLTLTA-----DEVAAALAQ--------HAEQRPLRQR----------LVA 430
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 152 MEEQCRDLNKRLERetlqskdfkleveklskrimalekLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKV---- 227
Cdd:PRK10246 431 LHGQIVPQQKRLAQ------------------------LQVAIQNVTQEQTQRNAALNEMRQRYKEKTQQLADVKTiceq 486
|
250
....*....|....*
gi 544346182 228 --RIKELEAIESRLE 240
Cdd:PRK10246 487 eaRIKDLEAQRAQLQ 501
|
|
| mukB |
PRK04863 |
chromosome partition protein MukB; |
17-239 |
3.05e-03 |
|
chromosome partition protein MukB;
Pssm-ID: 235316 [Multi-domain] Cd Length: 1486 Bit Score: 41.48 E-value: 3.05e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 17 RQKIQELTTNAKETHTKLALAEARVQEEEQkATRLEKELQTQTTKfHQDQDTIMAKLTNEDSQnRQLQQKLAALSRQIDE 96
Cdd:PRK04863 448 QAKEQEATEELLSLEQKLSVAQAAHSQFEQ-AYQLVRKIAGEVSR-SEAWDVARELLRRLREQ-RHLAEQLQQLRMRLSE 524
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 97 LEETNRSLRKAEEELQDIKEKISKGeYGNAgimAEVEELRkrvldmEGKDEELIKMEEQCRDLNKRLERETLQSKDFKLE 176
Cdd:PRK04863 525 LEQRLRQQQRAERLLAEFCKRLGKN-LDDE---DELEQLQ------EELEARLESLSESVSEARERRMALRQQLEQLQAR 594
|
170 180 190 200 210 220 230
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....
gi 544346182 177 VEKLSKRIM-------ALEKLEDAFN---KSKQECYSLKCN-LEKERmttkQLSQELESLKVRIKELEAIESRL 239
Cdd:PRK04863 595 IQRLAARAPawlaaqdALARLREQSGeefEDSQDVTEYMQQlLERER----ELTVERDELAARKQALDEEIERL 664
|
|
| EnvC |
COG4942 |
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, ... |
269-486 |
3.43e-03 |
|
Septal ring factor EnvC, activator of murein hydrolases AmiA and AmiB [Cell cycle control, cell division, chromosome partitioning];
Pssm-ID: 443969 [Multi-domain] Cd Length: 377 Bit Score: 40.90 E-value: 3.43e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 269 EKLKKTEDKLQAASSQLQVEQNKvttvTEKLIEETKRALKsktDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNML 348
Cdd:COG4942 23 AEAEAELEQLQQEIAELEKELAA----LKKEEKALLKQLA---ALERRIAALARRIRALEQELAALEAELAELEKEIAEL 95
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 349 KNRLQSLEAIEKDFLKN-KLNQDSGKSTTALHQENnkIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANER 427
Cdd:COG4942 96 RAELEAQKEELAELLRAlYRLGRQPPLALLLSPED--FLDAVRRLQYLKYLAPARREQAEELRADLAELAALRAELEAER 173
|
170 180 190 200 210
....*....|....*....|....*....|....*....|....*....|....*....
gi 544346182 428 DKAQFLSKELEHVKMELAKYKlAEKTETSHEqwLFKRLQEEEAKSGHLSREVDALKEKI 486
Cdd:COG4942 174 AELEALLAELEEERAALEALK-AERQKLLAR--LEKELAELAAELAELQQEAEELEALI 229
|
|
| SMC_prok_A |
TIGR02169 |
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of ... |
384-535 |
4.18e-03 |
|
chromosome segregation protein SMC, primarily archaeal type; SMC (structural maintenance of chromosomes) proteins bind DNA and act in organizing and segregating chromosomes for partition. SMC proteins are found in bacteria, archaea, and eukaryotes. It is found in a single copy and is homodimeric in prokaryotes, but six paralogs (excluded from this family) are found in eukarotes, where SMC proteins are heterodimeric. This family represents the SMC protein of archaea and a few bacteria (Aquifex, Synechocystis, etc); the SMC of other bacteria is described by TIGR02168. The N- and C-terminal domains of this protein are well conserved, but the central hinge region is skewed in composition and highly divergent. [Cellular processes, Cell division, DNA metabolism, Chromosome-associated proteins]
Pssm-ID: 274009 [Multi-domain] Cd Length: 1164 Bit Score: 40.82 E-value: 4.18e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 384 KIKELSQEVERL---KLKLKDMKAIEDDLMKTEdEYETLERRYANERDKAQfLSKELEHVKMELAKY--KLAEKTETSHE 458
Cdd:TIGR02169 192 IIDEKRQQLERLrreREKAERYQALLKEKREYE-GYELLKEKEALERQKEA-IERQLASLEEELEKLteEISELEKRLEE 269
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 459 qwLFKRLQEEEAKSGHL-SREVDALKEKIHEYMA----TEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKELER 533
Cdd:TIGR02169 270 --IEQLLEELNKKIKDLgEEEQLRVKEKIGELEAeiasLERSIAEKERELEDAEERLAKLEAEIDKLLAEIEELEREIEE 347
|
..
gi 544346182 534 YR 535
Cdd:TIGR02169 348 ER 349
|
|
| PTZ00108 |
PTZ00108 |
DNA topoisomerase 2-like protein; Provisional |
132-429 |
4.24e-03 |
|
DNA topoisomerase 2-like protein; Provisional
Pssm-ID: 240271 [Multi-domain] Cd Length: 1388 Bit Score: 41.19 E-value: 4.24e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 132 VEELRKRVL----DMEGKDEELIKMEEqcrDLNKRLERETLQSKDFKLEVEK------LSKRI--MALEKLEdafnkskq 199
Cdd:PTZ00108 1037 VKELKKLGYvrfkDIIKKKSEKITAEE---EEGAEEDDEADDEDDEEELGAAvsydylLSMPIwsLTKEKVE-------- 1105
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 200 ecyslkcNLEKERMTTKQLSQELESLKVR---IKELEAIESRLEKTEFTLKEDLTKLKTLTVM-FVDERKTMSEKLKKTE 275
Cdd:PTZ00108 1106 -------KLNAELEKKEKELEKLKNTTPKdmwLEDLDKFEEALEEQEEVEEKEIAKEQRLKSKtKGKASKLRKPKLKKKE 1178
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 276 DKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSL 355
Cdd:PTZ00108 1179 KKKKKSSADKSKKASVVGNSKRVDSDEKRKLDDKPDNKKSNSSGSDQEDDEEQKTKPKKSSVKRLKSKKNNSSKSSEDND 1258
|
250 260 270 280 290 300 310
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*...
gi 544346182 356 EAIEKDFLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKD--MKAIEDDLMKTEDEYE--TLERRYANERDK 429
Cdd:PTZ00108 1259 EFSSDDLSKEGKPKNAPKRVSAVQYSPPPPSKRPDGESNGGSKPSSptKKKVKKRLEGSLAALKkkKKSEKKTARKKK 1336
|
|
| CortBP2 |
pfam09727 |
Cortactin-binding protein-2; This entry is the first approximately 250 residues of ... |
147-265 |
5.33e-03 |
|
Cortactin-binding protein-2; This entry is the first approximately 250 residues of cortactin-binding protein 2. In addition to being a positional candidate for autism this protein is expressed at highest levels in the brain in humans. The human protein has six associated ankyrin repeat domains pfam00023 towards the C-terminus which act as protein-protein interaction domains.
Pssm-ID: 462860 [Multi-domain] Cd Length: 187 Bit Score: 39.12 E-value: 5.33e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 147 EELIKMEEQCRDLNKRLERETLqskdfklEVEKLSKRimALEKLEDafNKSKQECYSLK-----CNLEKERmttKQLSQE 221
Cdd:pfam09727 80 AELEKLVEKQRETQRRMLEQLA-------AAEKRHRR--VIRELEE--EKRKHARDTAQgddftYLLEKER---ERLKQE 145
|
90 100 110 120
....*....|....*....|....*....|....*....|....
gi 544346182 222 LESLKVRIKELEAiesRLEKTEFTLKEDLTKLKTLTVMFVDERK 265
Cdd:pfam09727 146 LEQEKAQQKRLEK---ELKKLLEKLEEELSKQKQIALLLVKERK 186
|
|
| HOOK |
pfam05622 |
HOOK protein coiled-coil region; This family consists of several HOOK1, 2 and 3 proteins from ... |
40-483 |
5.75e-03 |
|
HOOK protein coiled-coil region; This family consists of several HOOK1, 2 and 3 proteins from different eukaryotic organizms. The different members of the human gene family are HOOK1, HOOK2 and HOOK3. Different domains have been identified in the three human HOOK proteins, and it was demonstrated that the highly conserved NH2-domain mediates attachment to microtubules, whereas this central coiled-coil motif mediates homodimerization and the more divergent C-terminal domains are involved in binding to specific organelles (organelle-binding domains). It has been demonstrated that endogenous HOOK3 binds to Golgi membranes, whereas both HOOK1 and HOOK2 are localized to discrete but unidentified cellular structures. In mice the Hook1 gene is predominantly expressed in the testis. Hook1 function is necessary for the correct positioning of microtubular structures within the haploid germ cell. Disruption of Hook1 function in mice causes abnormal sperm head shape and fragile attachment of the flagellum to the sperm head. This entry includes the central coiled-coiled domain and the divergent C-terminal domain.
Pssm-ID: 461694 [Multi-domain] Cd Length: 528 Bit Score: 40.44 E-value: 5.75e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 40 RVQEEEQKATRLEKE---LQTQTTKFHQdqdtIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQdike 116
Cdd:pfam05622 15 RCHELDQQVSLLQEEknsLQQENKKLQE----RLDQLESGDDSGTPGGKKYLLLQKQLEQLQEENFRLETARDDYR---- 86
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 117 kiskgeygnagimAEVEELRKRVLDMEGKDEELIKMEEQCRDLNKRLE--RETlQSKDFKLEVEKLSKRimalEKLEDAF 194
Cdd:pfam05622 87 -------------IKCEELEKEVLELQHRNEELTSLAEEAQALKDEMDilRES-SDKVKKLEATVETYK----KKLEDLG 148
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 195 NKSKQ---------ECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIES----RLEKTEFTLKEDLTKLKTLTV--- 258
Cdd:pfam05622 149 DLRRQvklleernaEYMQRTLQLEEELKKANALRGQLETYKRQVQELHGKLSeeskKADKLEFEYKKLEEKLEALQKeke 228
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 259 MFVDERKTmsekLKKTEDKLQAAssqlQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKG 338
Cdd:pfam05622 229 RLIIERDT----LRETNEELRCA----QLQQAELSQADALLSPSSDPGDNLAAEIMPAEIREKLIRLQHENKMLRLGQEG 300
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 339 ND---------LLSRVNMLKNRLQ-----------SLEAIEKDFLKNKLNQ-----DSGKSTTALHQENNKIKELSQEVE 393
Cdd:pfam05622 301 SYrerltelqqLLEDANRRKNELEtqnrlanqrilELQQQVEELQKALQEQgskaeDSSLLKQKLEEHLEKLHEAQSELQ 380
|
410 420 430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 394 RLKLKLKD------------MKAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEhvkmelAKYKLAEKTETsheQWL 461
Cdd:pfam05622 381 KKKEQIEElepkqdsnlaqkIDELQEALRKKDEDMKAMEERYKKYVEKAKSVIKTLD------PKQNPASPPEI---QAL 451
|
490 500
....*....|....*....|..
gi 544346182 462 FKRLQEEEAKSGHLSREVDALK 483
Cdd:pfam05622 452 KNQLLEKDKKIEHLERDFEKSK 473
|
|
| COG1340 |
COG1340 |
Uncharacterized coiled-coil protein, contains DUF342 domain [Function unknown]; |
294-535 |
6.04e-03 |
|
Uncharacterized coiled-coil protein, contains DUF342 domain [Function unknown];
Pssm-ID: 440951 [Multi-domain] Cd Length: 297 Bit Score: 39.51 E-value: 6.04e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 294 TVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAiekdfLKNKLNQDSGK 373
Cdd:COG1340 1 SKTDELSSSLEELEEKIEELREEIEELKEKRDELNEELKELAEKRDELNAQVKELREEAQELRE-----KRDELNEKVKE 75
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 374 STTALHQENNKIKELSQEVERLKLKLKDMKAIEDDLMKTEDEYETLERRYANER----------DKAQFLSKELEHVKME 443
Cdd:COG1340 76 LKEERDELNEKLNELREELDELRKELAELNKAGGSIDKLRKEIERLEWRQQTEVlspeeekelvEKIKELEKELEKAKKA 155
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 444 LAKYKLAEKTETSHEQW------LFKRLQEEEAKSGHLSREVDALKEKIHEYMATEDLichLQGDHSVLQKKLNQQENRN 517
Cdd:COG1340 156 LEKNEKLKELRAELKELrkeaeeIHKKIKELAEEAQELHEEMIELYKEADELRKEADE---LHKEIVEAQEKADELHEEI 232
|
250
....*....|....*...
gi 544346182 518 RDLGREIENLTKELERYR 535
Cdd:COG1340 233 IELQKELRELRKELKKLR 250
|
|
| ClyA_Cry6Aa-like |
cd22656 |
Bacillus thuringiensis crystal 6Aa (Cry6Aa) toxin, and similar proteins; This model includes ... |
7-211 |
6.61e-03 |
|
Bacillus thuringiensis crystal 6Aa (Cry6Aa) toxin, and similar proteins; This model includes pesticidal Cry6Aa toxin from Bacillus thuringiensis, one of the many parasporal crystal (Cry) toxins produced during the sporulation phase of growth. Many of these proteins are toxic to numerous insect species and have been effectively used as proteinaceous insecticides to directly kill insect pests; some have been used to control insect growth on transgenic agricultural plants. Cry6Aa exists as a protoxin, which is activated by cleavage using trypsin. Structure studies for Cry6Aa support a mechanism of action by pore formation, similar to cytolysin A (ClyA)-type alpha pore-forming toxins (alpha-PFTs) such as HblB, and bioassay and mutation studies show that Cry6Aa is an active pore-forming toxin. Cry6Aa shows atypical features compared to other members of alpha-PFTs, including internal repeat sequences and small loop regions within major alpha helices.
Pssm-ID: 439154 [Multi-domain] Cd Length: 309 Bit Score: 39.66 E-value: 6.61e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 7 QRLTAQLTLQRQKIQELTTNAKETHTKLalaearvqeeeqkaTRLEKELQTQTTKFHQDQDTIMAKLTNEDSqnRQLQQK 86
Cdd:cd22656 117 KTIKALLDDLLKEAKKYQDKAAKVVDKL--------------TDFENQTEKDQTALETLEKALKDLLTDEGG--AIARKE 180
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 87 LAALSRQIDELEETNrsLRKAEEELQDIKEKISKGEygnagimAEVEELRKRVLDMEgkdeeliKMEEQCRDLNkrlere 166
Cdd:cd22656 181 IKDLQKELEKLNEEY--AAKLKAKIDELKALIADDE-------AKLAAALRLIADLT-------AADTDLDNLL------ 238
|
170 180 190 200
....*....|....*....|....*....|....*....|....*
gi 544346182 167 tlqskdfklevEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKE 211
Cdd:cd22656 239 -----------ALIGPAIPALEKLQGAWQAIATDLDSLKDLLEDD 272
|
|
| PRK00409 |
PRK00409 |
recombination and DNA strand exchange inhibitor protein; Reviewed |
20-120 |
7.56e-03 |
|
recombination and DNA strand exchange inhibitor protein; Reviewed
Pssm-ID: 234750 [Multi-domain] Cd Length: 782 Bit Score: 40.20 E-value: 7.56e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 20 IQELTTNAKETHTKLALAEARVQEeeqkATRLEKELQTQTTKFHQDQDTIMAKltnedsQNRQLQQKLAALSRQIDELEE 99
Cdd:PRK00409 522 IASLEELERELEQKAEEAEALLKE----AEKLKEELEEKKEKLQEEEDKLLEE------AEKEAQQAIKEAKKEADEIIK 591
|
90 100
....*....|....*....|....*..
gi 544346182 100 TNRSLRK------AEEELQDIKEKISK 120
Cdd:PRK00409 592 ELRQLQKggyasvKAHELIEARKRLNK 618
|
|
| CwlO1 |
COG3883 |
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function ... |
267-431 |
8.11e-03 |
|
Uncharacterized N-terminal coiled-coil domain of peptidoglycan hydrolase CwlO [Function unknown];
Pssm-ID: 443091 [Multi-domain] Cd Length: 379 Bit Score: 39.43 E-value: 8.11e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 267 MSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGN------- 339
Cdd:COG3883 28 LQAELEAAQAELDALQAELEELNEEYNELQAELEALQAEIDKLQAEIAEAEAEIEERREELGERARALYRSGGsvsyldv 107
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 340 --------DLLSRVNMLK----NRLQSLEAIEKDflKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIED 407
Cdd:COG3883 108 llgsesfsDFLDRLSALSkiadADADLLEELKAD--KAELEAKKAELEAKLAELEALKAELEAAKAELEAQQAEQEALLA 185
|
170 180
....*....|....*....|....
gi 544346182 408 DLMKTEDEYETLERRYANERDKAQ 431
Cdd:COG3883 186 QLSAEEAAAEAQLAELEAELAAAE 209
|
|
| Mplasa_alph_rch |
TIGR04523 |
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of ... |
96-532 |
8.66e-03 |
|
helix-rich Mycoplasma protein; Members of this family occur strictly within a subset of Mycoplasma species. Members average 750 amino acids in length, including signal peptide. Sequences are predicted (Jpred 3) to be almost entirely alpha-helical. These sequences show strong periodicity (consistent with long alpha helical structures) and low complexity rich in D,E,N,Q, and K. Genes encoding these proteins are often found in tandem. The function is unknown.
Pssm-ID: 275316 [Multi-domain] Cd Length: 745 Bit Score: 40.00 E-value: 8.66e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 96 ELEETNRSLRKAEEELQDIKEKISKGEYGNAGIMAEVEELRKRVLDMEGKdeeLIKMEEQCRDLNKRLERETLQSKDFKl 175
Cdd:TIGR04523 41 KLKTIKNELKNKEKELKNLDKNLNKDEEKINNSNNKIKILEQQIKDLNDK---LKKNKDKINKLNSDLSKINSEIKNDK- 116
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 176 evEKLSKRIMALEKLEDAFNKSKQECYSLKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLK- 254
Cdd:TIGR04523 117 --EQKNKLEVELNKLEKQKKENKKNIDKFLTEIKKKEKELEKLNNKYNDLKKQKEELENELNLLEKEKLNIQKNIDKIKn 194
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 255 -------TLTVMFVDERKTMS-----EKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTK 322
Cdd:TIGR04523 195 kllklelLLSNLKKKIQKNKSlesqiSELKKQNNQLKDNIEKKQQEINEKTTEISNTQTQLNQLKDEQNKIKKQLSEKQK 274
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 323 ERDDLKNKLKAEEEKGNDLLSRVNMLKN-RLQSLEAIEKDFLKNKLNQDSgKSTTALHQENNKIKELSQEVERLKLKLKD 401
Cdd:TIGR04523 275 ELEQNNKKIKELEKQLNQLKSEISDLNNqKEQDWNKELKSELKNQEKKLE-EIQNQISQNNKIISQLNEQISQLKKELTN 353
|
330 340 350 360 370 380 390 400
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 402 M----KAIEDDLMKTEDEYETLERRYANERDKAQFLSKELEHVKMELAKYKLAE-------KTETSHEQWLFKRLQEEEA 470
Cdd:TIGR04523 354 SesenSEKQRELEEKQNEIEKLKKENQSYKQEIKNLESQINDLESKIQNQEKLNqqkdeqiKKLQQEKELLEKEIERLKE 433
|
410 420 430 440 450 460
....*....|....*....|....*....|....*....|....*....|....*....|....*.
gi 544346182 471 KSGHLSREVDALKEKIHE----YMATEDLICHLQGDHSVLQKKLNQQENRNRDLGREIENLTKELE 532
Cdd:TIGR04523 434 TIIKNNSEIKDLTNQDSVkeliIKNLDNTRESLETQLKVLSRSINKIKQNLEQKQKELKSKEKELK 499
|
|
| PRK09039 |
PRK09039 |
peptidoglycan -binding protein; |
7-137 |
9.00e-03 |
|
peptidoglycan -binding protein;
Pssm-ID: 181619 [Multi-domain] Cd Length: 343 Bit Score: 39.18 E-value: 9.00e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 7 QRLTAQLTLQRQKIQELTTNAKETHTKLALAEA---RVQEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQL 83
Cdd:PRK09039 63 AELADLLSLERQGNQDLQDSVANLRASLSAAEAersRLQALLAELAGAGAAAEGRAGELAQELDSEKQVSARALAQVELL 142
|
90 100 110 120 130
....*....|....*....|....*....|....*....|....*....|....*
gi 544346182 84 QQKLAALSRQIDELEEtnrSLRKAEEELQDIKEKISK-GEYGNAGIMAEVEELRK 137
Cdd:PRK09039 143 NQQIAALRRQLAALEA---ALDASEKRDRESQAKIADlGRRLNVALAQRVQELNR 194
|
|
| PspC_subgroup_1 |
NF033838 |
pneumococcal surface protein PspC, choline-binding form; The pneumococcal surface protein PspC, ... |
76-431 |
9.09e-03 |
|
pneumococcal surface protein PspC, choline-binding form; The pneumococcal surface protein PspC, as described in Streptococcus pneumoniae, is a repetitive and highly variable protein, recognized by a conserved N-terminal domain and also by genomic location. This form, subgroup 1, has variable numbers of a choline-binding repeat in the C-terminal region, and is also known as choline-binding protein A. The other form, subgroup 2, is anchored covalently after cleavage by sortase at a C-terminal LPXTG site.
Pssm-ID: 468201 [Multi-domain] Cd Length: 684 Bit Score: 39.61 E-value: 9.09e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 76 EDSQNRQLQQKLAALSRQIdeLEETNRSLRKAEEEL-QDIKEKISkgeygnagimAEVEELRKRVLDMEGKDEELIKMEE 154
Cdd:NF033838 82 KHTQNVALNKKLSDIKTEY--LYELNVLKEKSEAELtSKTKKELD----------AAFEQFKKDTLEPGKKVAEATKKVE 149
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 155 QCRDLNKRLERE------TLQSKDFKLEVEKLSKRImalekledafNKSKQECYSLKCNLEKERMTTKQLSQELESLKVR 228
Cdd:NF033838 150 EAEKKAKDQKEEdrrnypTNTYKTLELEIAESDVEV----------KKAELELVKEEAKEPRDEEKIKQAKAKVESKKAE 219
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 229 IKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAASSQLQVEQNKVTTVTEKLIEETKRALK 308
Cdd:NF033838 220 ATRLEKIKTDREKAEEEAKRRADAKLKEAVEKNVATSEQDKPKRRAKRGVLGEPATPDKKENDAKSSDSSVGEETLPSPS 299
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 309 SKTdvEEKMYSVTKERDDLKNKLKAEEEKgndllSRVNMLKNRLQSL--EAIEKDFLKNKLNQDSGKSTTALHQENNKIK 386
Cdd:NF033838 300 LKP--EKKVAEAEKKVEEAKKKAKDQKEE-----DRRNYPTNTYKTLelEIAESDVKVKEAELELVKEEAKEPRNEEKIK 372
|
330 340 350 360
....*....|....*....|....*....|....*....|....*
gi 544346182 387 ELSQEVERLKLKLKDMKAIEDDLMKTEDEyetlERRYANERDKAQ 431
Cdd:NF033838 373 QAKAKVESKKAEATRLEKIKTDRKKAEEE----AKRKAAEEDKVK 413
|
|
| COG4372 |
COG4372 |
Uncharacterized protein, contains DUF3084 domain [Function unknown]; |
42-406 |
9.12e-03 |
|
Uncharacterized protein, contains DUF3084 domain [Function unknown];
Pssm-ID: 443500 [Multi-domain] Cd Length: 370 Bit Score: 39.50 E-value: 9.12e-03
10 20 30 40 50 60 70 80
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 42 QEEEQKATRLEKELQTQTTKFHQDQDTIMAKLTNEDSQNRQLQQKLAALSRQIDELEETNRSLRKAEEELQDIKEKISKG 121
Cdd:COG4372 6 EKVGKARLSLFGLRPKTGILIAALSEQLRKALFELDKLQEELEQLREELEQAREELEQLEEELEQARSELEQLEEELEEL 85
|
90 100 110 120 130 140 150 160
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 122 EygnagimaevEELRKRVLDMEGKDEELIKMEEQCRDLNKRLERETLQSKDFKLEVEKLSKRIMALEKLEDAFNKSKQEC 201
Cdd:COG4372 86 N----------EQLQAAQAELAQAQEELESLQEEAEELQEELEELQKERQDLEQQRKQLEAQIAELQSEIAEREEELKEL 155
|
170 180 190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 202 YSlKCNLEKERMTTKQLSQELESLKVRIKELEAIESRLEKTEFTLKEDLTKLKTLTVMFVDERKTMSEKLKKTEDKLQAA 281
Cdd:COG4372 156 EE-QLESLQEELAALEQELQALSEAEAEQALDELLKEANRNAEKEEELAEAEKLIESLPRELAEELLEAKDSLEAKLGLA 234
|
250 260 270 280 290 300 310 320
....*....|....*....|....*....|....*....|....*....|....*....|....*....|....*....|
gi 544346182 282 SSQLQVEQNKVTTVTEKLIEETKRALKSKTDVEEKMYSVTKERDDLKNKLKAEEEKGNDLLSRVNMLKNRLQSLEAIEKD 361
Cdd:COG4372 235 LSALLDALELEEDKEELLEEVILKEIEELELAILVEKDTEEEELEIAALELEALEEAALELKLLALLLNLAALSLIGALE 314
|
330 340 350 360
....*....|....*....|....*....|....*....|....*
gi 544346182 362 FLKNKLNQDSGKSTTALHQENNKIKELSQEVERLKLKLKDMKAIE 406
Cdd:COG4372 315 DALLAALLELAKKLELALAILLAELADLLQLLLVGLLDNDVLELL 359
|
|
| |